Understanding donor screening rules for human cell or tissue products
Guidance on TGO 108: Requirements around the minimisation of transmission of communicable diseases for therapeutic goods containing biologicals and human cell or tissue (HCT).
Purpose
This guidance is for manufacturers and sponsors of biologicals and human cell or tissue (HCT) products. It specifies requirements around the minimisation of transmission of communicable diseases through the use of therapeutic goods comprising, derived from or containing HCT, as set out in the Therapeutic Goods (Standard for Human Cell and Tissue Products – Donor Screening Requirements) (TGO 108) Order 2021, which came into effect 30 September 2021.
Therapeutic Goods Orders (TGOs) are standards that determine the consistency of product quality, including label quality. As a provider of such therapeutic goods, you must comply with requirements that contribute to their quality and safety, and that mitigate infectious disease risks.
TGO 108 replaces:
TGO 88 was due to ‘sunset’ on 1 October 2023. Sunsetting is the process whereby legislative instruments undergo automatic repeal after 10 years following their registration.
TGA updated TGO 88 at the same time as numerous other TGOs which sunsetted on 1 October 2021 because all these TGOs were inter-related and cross-referenced each other:
- Therapeutic Goods Order No. 83 - Standards for human musculoskeletal tissue (TGO 83)
- Therapeutic Goods Order No. 84 - Standards for human cardiovascular tissue (TGO 84)
- Therapeutic Goods Order No. 85 - Standards for human ocular tissue (TGO 85)
- Therapeutic Goods Order No. 86 - Standards for human skin (TGO 86).
This information is provided for guidance only and has been developed based on current knowledge of the subject matter.
It should not be relied on to address every aspect of the relevant legislation. You should seek your own independent legal advice to ensure that all legislative requirements are met.
Please read this guidance in conjunction with TGO 108.
About TGO 108
The Therapeutic Goods (Standard for Human Cell and Tissue Products – Donor Screening Requirements) (TGO 108) Order 2021 specifies the minimum criteria and screening requirements with regard to donor:
- medical and social history requirements
- testing requirements for blood and other samples
- physical assessment requirements
- deferral criteria.
Additional or higher requirements may be included at a sponsor’s discretion or may be required by a product-specific order or default standard. Further testing as clinically relevant for specific products or patient populations may also be necessary, for example, Cytomegalovirus testing where recipients are children or immunocompromised.
TGA accepts that alternative approaches may be suitable to satisfy the requirements of TGO 108 for specific product types, provided there is appropriate justification. Where alternative approaches are used, these must be validated by the manufacturer.
The justification and supporting validation information is subject to TGA review and approval. It must be provided in the data supporting the application for product approval or in an application for a variation, post approval. Subject to approval, and requirements in the standard, sponsor may still be required to obtain consent prior to import, supply or export therapeutic goods that do not comply with a standard applicable to the goods, see section 14 the Therapeutic Goods Act 1989 (the Act).
TGO 108 applies to:
- biologicals (encompassing starting materials and finished therapeutic goods) comprising, derived from, or containing HCTs
- all human donors of one or more of the following that are collected from a donor for use in the manufacture of an HCT product:
- human blood and blood components including:
- red cells
- white cells
- platelets and
- plasma (including plasma for fractionation)
- human cells (including haematopoietic progenitor cells).
TGO 108 does not apply to biologicals that are:
- not intended for therapeutic use, that is, blood or tissue samples for infectious disease or bioburden testing
- excluded from TGA regulation
- faecal microbiota transplant (FMT) products (also see TGO 105)
- autologous HCT products.
Although autologous HCT products are exempt from TGO 108, any or all of the requirements specified in TGO 108 could be required for demonstrating compliance to TGO 109.
Autologous HCT products have a lower risk of transmitting infectious diseases than their allogeneic counterparts, there are nonetheless risks associated with contamination or cross-contamination of such products or other products during manufacture. In order to efficiently and effectively mitigate these risks, the following regulatory requirements must be met:
In the case of ‘autologous HCT products regulated with exemptions’:1
- These products can only be:
- minimally manipulated, and
- deployed for homologous use
- for a single indication in a single clinical procedure
- manufactured and administered under high-level clinical oversight
- These criteria clarify the boundaries between clinical practice, which is already regulated to manage such risks, and therapeutic goods regulations.
- TGA requirements such as adverse event reporting and recalls continue to apply. Thus TGO 107 and TGO 109 also do not apply to these products.
In the case of fully regulated autologous HCT products:
- To consider this risk, the remade TGO 109 includes a section titled: ‘Diseases and conditions that may compromise biologicals’. This section clarifies that where a risk of contamination or cross-contamination of other products manufactured at the same facility (amongst other factors that may affect the quality, safety or efficacy) is possible, any or all the donor screening requirements specified in TGO 108 can be applied.
- Further, the requirements of donor traceability will continue to apply to such products via TGO 107.
- GMP requirements also apply to these products.
GMP requirements continue to apply for autologous HCT products that do not meet the ‘autologous HCT products regulated with exemptions’ criteria. These GMP requirements include measures for the minimisation of contamination or cross-contamination of premises, equipment, and of any other therapeutic goods that are manufactured at the shared facility. Other areas that need to be considered are personnel working in areas where contamination may be an issue (for example, clean areas or areas where infectious materials are handled) – such personnel must be given specific training. For further information, refer to:
Review of TGO 108
TGO 108 will be reviewed regularly as changes in legislation, emerging technology, and best practice occur. Sponsors and manufacturers are encouraged to discuss with TGA any proposed changes in practice or evolving technologies that may affect, or be affected by, the requirements of TGO 108.
Part 1 – Preliminary
Section 4 - Definitions
The definitions provided within TGO 108 are specific to this TGO and are not intended to apply outside of TGO 108. Please see TGO 108 for definitions.
Where possible, definitions included in TGA’s Acronyms and glossary terms are derived from current TGA legislation, international regulatory documents, or current industry guidance documents. Where a suitable definition has not been identified by those means, TGA has developed a definition that is informed by equivalent terms used in the aforementioned documents, expert committees, or through public consultation.
Further clarification of some definitions, specific to TGO 108, are provided below.
allogeneic use, in relation to an HCT product, means administration to, or application in the treatment of, a person other than the person from whom the HCT materials used in the manufacture of the product were collected.
This definition intends to capture all non-self-transplantation or transfusion, and includes syngeneic use (for example, between identical twins).
asystole, in relation to a donor of HCT materials, means the reference time for cardiac arrest, which is determined as follows:
- the documented pronounced time of death; or
- if death is not witnessed – the last time the donor was known to be alive; or
the tissue donor is also a solid organ donor – the cross-clamp time.
This definition includes cross-clamp time, which for organ donors refers to the time that the aorta is cross clamped by a surgical team.
A documented pronounced time of death is used as asystole when life-saving procedures have been attempted and there were signs of, or documentation of, recent life (for example, agonal respirations, pulse-less electrical activity).
The time of certification of circulatory death would be considered appropriate if the tissue donor is also a solid organ donor.
HCT materials means one or more of the following that are collected from a donor for use in the manufacture of an HCT product:
- human cells (including haematopoietic progenitor cells);
- human tissues;
- blood;
- blood components (including plasma).
HCT materials applies to collected material and at any stage during processing for manufacture of HCT product. Note that the blood component definition is not an inclusive list.
Part 2 – Requirements
Part 2 specifies the minimum standards for samples and test methods used for infectious disease testing for determining donor suitability.
Section 9 - General requirements
Subsections 9(1) to 9(3) - Manufacture
9 General requirements
- An HCT product must be manufactured in accordance with procedures that mitigate the risk of infectious disease transmission.
- An HCT product must not be released for supply, unless the applicable screening procedures and requirements specified in this instrument are satisfied.
- Acceptance criteria based on microbial specifications must be applied to HCT materials used in the manufacture of an HCT product.
All manufacturers must ensure that the design and construction of the manufacturing facility is suitable to the type of processing conducted, with separation of areas for minimising cross- contamination.
Design and develop the facility and procedures, taking into account the principles of Good Manufacturing Practice (GMP) if the manufacture of an HCT product takes place in a hospital.
Where a facility holds a TGA manufacturing licence, complying with the requirements of the Australian Code of GMP achieves compliance with the requirements outlined in this subsection. Guidance is available to assist facilities on how the Australian Code of GMP applies to the manufacture of HCT products.
All HCT products and materials must be segregated from the mainstream inventory until the HCT product has been determined to be compliant with all necessary requirements or release criteria. This is especially relevant for products stored in an inventory (or banked) with other products for potentially long periods of time. These products pose a risk of transmission to other products through physical contact, and a risk to the public from unintentional release and use.
Quarantine procedures are assessable under the current Good Manufacturing Practice (cGMP) requirements. Conditions of quarantine should be consistent with storage conditions, wherever possible.
Subsection 9(4) - Blood sample testing timeframe
9 General requirements
- Blood samples of a donor of HCT materials must be tested:
- as soon as practicable after the blood sample is taken; or
- in accordance with the timeframe specified by the manufacturer of the IVD medical device or the in-house IVD medical device that is used for testing the sample; or
- where testing is conducted outside of Australia – within a timeframe that is validated by the testing laboratory.
Ensure timeframes for sample testing are in accordance with requirements of validated test kits. Validate testing methodology for cadaveric samples to include the relevant time period of sample collection following asystole.
Subsection 9(5) - Blood sample testing using IVDs
9 General requirements
- Blood samples must be tested using IVD medical devices or in-house IVD medical devices that:
- use the most appropriate methodology available that is validated for testing the samples (including cadaveric samples); and
- where testing is conducted in Australia – are either included in the Register or exempt from the requirement to be included in the Register, or are the subject of an approval or authority under the Act; and
- where testing is conducted outside of Australia – are:
- used in a facility that has been approved for such testing by a relevant regulatory authority in the country in which the testing is conducted; and
- considered acceptable by the Therapeutic Goods Administration.
Each in vitro diagnostic (IVD) medical device or in-house IVD medical device used for the mandatory donor screening tests and confirmatory tests (if they support final product release) should be:
- validated for the purpose for which it is to be used (intended use)
- used in accordance with the test kit instructions.
Where test methods are used beyond the test kit instructions for example, use on cadaveric samples), validation to support the extended use must be demonstrated.
Methodology of testing kits
Determine and justify the most appropriate methodology. The infectious diseases test screening protocol could be an ‘in-house’ test or a commercial kit and may be conducted by the sponsor or a contract laboratory. Consider utilising new test methods, such as more sensitive assays, as they become available.
When testing a sample of cadaveric blood, the screening test must be specifically approved for use on cadaveric specimens or validated for this purpose by the testing laboratory.
Approval of test kits
Where test methods are used beyond the level approved by the local regulatory approval, for example, use on cadaveric samples, validation to support the extended use must be demonstrated.
A well-characterised assay already established for blood donors may be used for testing cadaveric specimens. Alternative methods to characterise a cadaveric assay may be acceptable to TGA and the supporting validation data should be provided for review with the application.
Testing facility
All facilities performing donor testing must be approved by the regulatory authority in the country where the testing is performed.
For domestic facilities, a TGA licence will be required.
For overseas testing facilities, the sponsor must hold a TGA clearance demonstrating compliance with GMP requirements. Where confirmatory testing is utilised to decide suitability of a donor, the facility’s GMP must include this purpose.
Acceptance of testing kits
Sponsors are encouraged to contact TGA’s Biological Science Section (BSS) as to the acceptability of test kits.
Test kits used domestically for donor screening should meet the requirements of the IVD regulatory framework. Testing facilities in Australia would also need a GMP licence from TGA.
For test kits used overseas, sponsors are encouraged to contact TGA’s Biological Science Section (BSS) as to kit acceptability.
Subsection 9(6) – Blood sample testing by laboratories
9 General requirements
- Where the testing of a blood sample is conducted by a laboratory that is not under the direct control of the manufacturer of the relevant HCT product:
- the testing must be conducted under a contract between the manufacturer and the laboratory; and
- the contract mentioned in paragraph (a) must clearly set out the responsibilities of the manufacturer and the laboratory, and include arrangements to ensure that information relating to matters in this section and any other relevant details relating to the IVD medical devices or in house IVD medical devices used for such testing can be obtained from the laboratory.
When external laboratories are used for testing of blood samples, a contract must be in place to stipulate the responsibilities of the manufacturer. The contract should clearly outline:
- what tests the laboratory will do
- the IVDs that they will use
- the record keeping or reporting structure in place
- the responsibility of the testing laboratory to notify the manufacturer of any changes to IVDs.
Subsections 9(7) and 9(8) – Plasma dilution
9 General requirements
- The testing of blood samples must take into account all factors that may cause plasma dilution.
- Where plasma dilution is suspected at a level sufficient to alter the test results in relation to a blood sample, and a pre-infusion sample is unavailable for testing, then:
- an algorithm must be applied to assess the extent of plasma dilution; and
- the extent of plasma dilution must be less than 50 per cent, unless use of samples with more than 50 per cent plasma dilution is validated by the manufacturer of the IVD medical devices or in-house IVD medical devices used for testing the sample.
Consider all factors that may cause plasma dilution when testing of blood samples. The results of infectious disease testing may be affected where a potential donor may have recently received an intravenous treatment, for example infusion of blood components, colloids, or crystalloids.
Undertake a complete haemodilution assessment on all living and deceased donors for infectious disease test screening protocols. Plasma dilution algorithms may be used to calculate appropriate amounts.
If the infusion or transfusion volume for the donor totals more than 2000 mL, it is considered essential that a haemodilution assessment be performed if:
- an infusion or transfusion of blood or colloids was received within 48 hours preceding collection of the sample or within 48 hours preceding death, whichever occurred earlier
- an infusion of crystalloids was received within one hour preceding collection of the sample, or within one hour preceding death, whichever occurred earlier
- a combination of blood, crystalloids or colloids was received within the applicable timeframes.
It is essential that an assessment be performed where there have been any infusions or transfusions that might affect test results in a child of less than 12 years of age. The time frames are as defined above.
Record the date and time of sample collection.
An appropriate algorithm must be used to evaluate that plasma dilution and show that it is not sufficient to affect the test results. The plasma dilution factor should be less than 50 per cent, that is, if greater than 50 per cent dilution has occurred, then the post-transfusion/infusion specimen should not be used.
When performing the algorithm:
- consider volumes of medications administered with IV fluids
- if the volume administered is unknown, consider the worst-case scenario that all was given.
Subsections 9(9) and 9(10) – Record keeping
9 General requirements
- Records must be maintained in relation to the following:
- the tests performed in relation to blood samples and the results of those tests; and
- the IVD medical device or in-house IVD medical device used for testing the samples; and
- any test modifications; and
- any evaluations of, or anomalies in, test results.
- Procedures must be implemented for notifying a donor of HCT materials, a relevant health practitioner, a relevant hospital and any other relevant organisation, of a test result in relation to the donor that is indicative of a disease or carrier state.
The retention of records is required by TGO 108 and is consistent with the requirements of the current GMP. Records can be kept in paper or electronic form and must be available to GMP inspectors.
The period of retention must take into account jurisdictional or hospital policies. The application should specify and justify record retention periods and include consideration of product risk, shelf life of product, and timeframe for which the product is expected to have a therapeutic or physiological function in the recipient. A documented process must be in place to demonstrate that donor test results that are indicative of an infectious disease or carrier state are reported back to that donor, where necessary.
Section 10 - Medical and social history of donors
Section 10 specifies the minimum standards for assessing the suitability of a donor. The medical and social history is the first tier of risk minimisation in terms of infectious disease transmission since high risk donors can be identified before donation, or, where allowed, after donation but before introduction of the therapeutic good into the mainstream inventory of the manufacturing facility.
Subsections 10(1) and 10(2) - Donor interviews
10 Medical and social history of donors
Living donors
- A medical and social history in relation to a living donor of HCT materials, covering the ineligibility criteria for donor selection specified in Schedule 1 and any other relevant matters, must be obtained by interview.
- The interview must be:
- conducted by an interviewer who is:
- appropriately qualified and trained; and
- an employee of, or under a contract with, a person engaged in the collection of HCT materials or the manufacture of the HCT product; and
- held face-to-face (to the extent that it is possible in the circumstances) with the donor or the donor’s guardian or next of kin; and
- conducted within 30 days before or 30 days after the collection of the HCT materials; and
- documented.
For living donors, a face-to-face interview with the donor or the donor’s guardian or next of kin is preferred, but it is recognised that this may not be possible within the specified timeframe. Describe the alternative process where a face-to-face interview is not conducted.
Obtaining preliminary information without a trained interviewer prior to interview may be acceptable. Confirmation of information/currency of donor history must be in the presence of a trained interviewer within 30 days before or 30 days after the collection and must be completed before HCT product is released from quarantine. To ensure the relevance of the information collected, the timeframes around when the donor interview is performed should be as close to the collection date as feasible.
Subsection 10(3) - Deceased donor: medical and social history
10 Medical and social history of donors
Deceased donors
- A medical and social history in relation to a deceased donor of HCT materials, covering the ineligibility criteria for donor selection specified in Schedule 1 and any other relevant matters, must be obtained and documented within 7 days before or 7 days after the collection of the HCT materials, by:
- both:
- an interview with a person who is sufficiently informed about the donor’s medical and social history; and
- an examination of relevant documentation in relation to the donor; or
where the interview mentioned in subparagraph (3)(a)(i) is not possible—an examination of the donor documentation to ensure there is sufficient evidence to determine the acceptability of the donor’s medical and social history.
Example: A person who is sufficiently informed about a deceased donor’s medical and social history may include the donor’s treating physician, next of kin or closest available relative, a member of the donor’s household, or a person with a relationship with the donor, such as a carer, friend or partner.
For deceased donors, it is recognised that the next of kin (sufficiently informed person) may not be available for interview within the required timeframe. In this situation, an examination of the donor documentation to ensure there is sufficient evidence to determine the acceptability of the donor’s medical and social history may provide the necessary evidence to obtain the donor history within the specified timeframe.
Subsection 10(4) and 10(5) – Donors of HCT materials for plasma fractionation: medical and social history
10 Medical and social history of donors
Donors of HCT materials used exclusively for plasma fractionation
- The periods of ineligibility specified in column 3 of items 2, 4, 13, 16 to 19, and 23 to 26 of the table in Schedule 1 do not apply in relation to a donor of HCT materials that are used exclusively for plasma fractionation in the manufacture of HCT products.
- The periods of ineligibility specified in column 3 of items 1 to 7 and column 3 of items 9 to 26 of the table in Schedule 1 do not apply in relation to a donor of HCT materials that are used exclusively for plasma fractionation in the manufacture of an export only medicine.
This subsection provides specific exemption from certain donor history criteria and deferrals for plasma for fractionation.
‘Plasma for fractionation’ is defined in European Pharmacopoeia (Ph. Eur.) monograph 0853 as the liquid part of human blood remaining after the separation of the cellular elements from blood collected and is intended for the manufacture of plasma-derived products.
The only period of ineligibility that applies to ‘HCT materials that are used exclusively for plasma fractionation in the manufacture of an export only medicine’ is specified in column 3 of item 8 of the table Schedule 1.
Subsection 10(6) – Ocular donors: medical and social history
10 Medical and social history of donors
Donors of HCT materials that are human ocular tissue only
- The periods of ineligibility specified in column 3 of items 16 to 19 of the table in Schedule 1 do not apply in relation to a donor of HCT materials that are human ocular tissue only.
This subsection provides specific exemption from certain donor history criteria and deferrals for donors of ocular tissue.
Subsection 10(7) – Change in circumstances of donor
10 Medical and social history of donors
Change of circumstance of donor
- Where the circumstances of a donor of HCT materials used in the manufacture of an HCT product change in relation to the ineligibility criteria for donor selection specified in Schedule 1, the relevant aspects of the medical and social history of the donor must be reviewed by the manufacturer of the HCT product with respect to those changes, before the HCT product is released for supply.
A change in circumstances for a donor of HCT may include detection of a latent condition after donation or previous travel to an area that has been found to have an outbreak of a disease (for example, malaria, Dengue fever, COVID-19). In these instances, donor eligibility should be reviewed prior to release of HCT product.
Subsections 10(8) and 10(9) – Screening requirements
10 Medical and social history of donors
Screening requirements
- An HCT product must not be released for supply, unless the medical and social history of a donor is reviewed and evaluated in accordance with this section.
- Where a donor meets any of the medical and social history criteria specified in column 2 of an item of the table in Schedule 1, the donor must be subjected to the period of ineligibility specified in column 3 of that item in relation to the collection of HCT materials from the donor for use in the manufacture of HCT products.
Where possible and practicable these criteria and deferral periods are harmonised with international criteria.
Subsections 10(10) to 10(13) – Vertical transmission risk
10 Medical and social history of donors
Screening in relation to donors less than 18 months old
- If the donor is less than 18 months old, a medical and social history in relation to the donor’s birth mother, covering the ineligibility criteria for donor selection specified in column 2 of items 1 to 7, 11 to 13, 17 and 20 of the table in Schedule 1 must be obtained.
- Where the donor’s birth mother meets the medical and social history criteria specified in column 2 of item 1 to 7, 11 to 13, 17 or 20 of the table in Schedule 1, then the donor must be subjected to the period of ineligibility specified in column 3 of that item, in relation to the collection of HCT materials from the donor.
Screening in relation to donors who have consumed breast milk.- If the donor has consumed breast milk from a person (the relevant person) within the previous six months, a medical and social history in relation to the relevant person, covering the ineligibility criteria for donor selection specified in column 2 of the items 1 to 7, 11 to 13, 17 and 20 of the table in Schedule 1 must be obtained.
- Where the relevant person meets the medical and social history criteria specified in column 2 of item 1 to 7, 11 to 13, 17 or 20 of the table in Schedule 1, then the donor must be subjected to the period of ineligibility specified in column 3 of that item, in relation to the collection of HCT materials from the donor.
This subsection provides specific criteria that apply to the mothers of infant donors or the relevant persons to reduce the risk of vertical transmission of disease via placenta or lactation. This clause also applies to HCT material donor who has consumed breast milk.
Subsection 10(14) – Directed allogeneic donation
10 Medical and social history of donors
HCT products that are manufactured for directed allogeneic use
- Subsections (9), (11) and (13) do not apply in relation to HCT products that are manufactured for directed allogeneic use where the medical or dental practitioner who is responsible for the administration to, or application in the treatment of, the designated patient is provided the complete medical and social history of the donor of the HCT materials, including (where applicable) the medical and social history of the donor’s birth mother or the relevant person.
In certain circumstances, subsections (9), (11) and (13) do not apply to HCT products that are manufactured for directed allogeneic use.
Section 11 - Blood samples - taking and testing
Section 11 specifies the minimum standards for samples and test methods used for infectious disease testing. Quality of the samples and validation of the methods used in donor testing is critical for determining donor suitability.
Subsections 11(1) to 11(4) - Living donor blood sampling
11 Blood samples – taking and testing
- Blood samples must be:
- taken from a donor of HCT materials; and
- tested for the purpose of donor screening.
- Blood samples must be taken using aseptic procedures.
Blood samples – living donors of HCT materials other than materials used exclusively for plasma fractionation
- Subject to subsections (4) and (5), blood samples of a living donor of HCT materials must:
- be taken within 7 days before, or 7 days after, the collection of the HCT materials from the donor; and
- undergo both NAT and serology testing in accordance subsection (7).
- Where an HCT product is able to be stored for more than 180 days without compromising the quality, safety or efficacy of the product, blood samples of a living donor of the HCT materials used in the manufacture of the product may instead:
- be taken:
- within 7 days before, or 7 days after, the collection of the HCT materials from the donor; and
- at least 180 days after the collection of the HCT materials from the donor; and
- undergo serology testing in accordance with subsections (8) and (9).
The 7-day window for living donors represents the nominal timeframe to collect samples to accurately reflect the donor infectious disease status. It may be appropriate for manufacturers to apply more stringent timeframes to some cell or tissue types, such as fresh blood.
Subsection 11(5) – Blood sampling for plasma fractionation
11 Blood samples – taking and testing
Blood samples – living donors of HCT materials used exclusively for plasma fractionation
- Blood samples of a living donor of HCT materials that are used exclusively for plasma fractionation must:
- be taken within 7 days before, or 7 days after, the collection of the HCT materials from the donor; and
- undergo both NAT and serology testing in accordance subsection (11).
Where the HCT materials donated solely for the purpose for plasma fractionation, such donors are not required to undergo testing for HTLV or syphilis. All other tests specified in subsection 11(11) must be non-reactive.
The term ‘non-reactive’ is intended to include other terminology for the same test result such as ‘negative’ and ‘not detected’.
Justify the decision as to which test results lead to a ‘reactive’ or ‘non-reactive’ test. In addition, justify the role of any confirmatory testing in the determination of meeting this requirement.
Subsection 11(6) - Deceased donor blood sampling
11 Blood samples – taking and testing
Blood samples – deceased donors of HCT materials
- Blood samples of a deceased donor of HCT materials must:
- be taken:
- in accordance with subsection 9(4); or
- within 7 days before the collection of the HCT materials; and
- undergo NAT and serology testing in accordance with subsection (7).
The quality of the cadaveric blood sample must be suitable to allow infectious disease testing to be performed. Instructions for use provided with a test method may define the appropriate time frame for collection of the blood sample prior to testing, or the appropriate time frame should be validated.
Subsections 11(7) to 11(10) – Living and deceased donor infectious disease testing
11 Blood samples – taking and testing
Testing – living and deceased donors of HCT materials
- Subject to subsection (9), the following testing must be conducted in relation to blood samples taken in accordance with paragraphs (3)(a) or (6)(a):
- serology testing for HIV-1, HIV-2, HCV, HBsAg, HTLV-1, HTLV-2, and syphilis (Treponema pallidum); and
- NAT for HIV-1, HIV-2, HBV, and HCV.
- Subject to subsection (9), the following testing must be conducted in relation to blood samples taken in accordance with subparagraph (4)(a)(i):
- serology testing for HIV-1, HIV-2, HCV, HBsAg, HTLV-1, HTLV-2, and syphilis (Treponema pallidum).
- Serology testing for HTLV-1 and HTLV-2 is not required if a risk assessment, in relation to the type of the HCT materials and the geographical history of the donor, is undertaken, which demonstrates that:
- the risk of transmission of HTLV-1 and HTLV-2 from the donor is mitigated in the absence of donor testing; and
- the donor, the parents of the donor, and the sexual partners of the donor, originate from a low risk area; and
- the HCT materials is not a viable leukocyte-rich cell or tissue.
- The following testing must be conducted in relation to blood samples taken in accordance with subparagraph (4)(a)(ii):
- serology testing for HIV-1, HIV-2, HCV, and HBsAg.
Document the policy for determining the individual infectious disease status based on test results. For example, HBV testing algorithms required to interpret the status of the donor (whether NAT positive/serology negative result or NAT negative/serology positive result).
If the test result for syphilis is reactive by a non-specific test, there are two options:
- discard product
- perform a specific confirmatory test to rule out the positive result from the non-specific test (non-specific [reaginic] syphilis tests are prone to false positive results) Undertake initial serology and, where applicable, NAT on the sample collected at the time of donation during the timeframe specified.
There are two main infectious diseases testing options:
- for HCT, the mandatory requirements for initial sample serology as well as its initial sample NAT for HIV, HCV and HBV can be performed
- for HCT that can be stored for over 180 days, initial sample serology is mandatory, and the donor must be sampled and tested by serology again more than 180 days after HCT collection.
If serology testing is not able to be done after 180 days (as described above), it may be possible to justify retrospective testing of the initial donor sample by NAT in combination with the initial sample serology. Demonstrate the validity/suitability of using donor samples that may have been held in longer-term storage prior to undertaking NAT on the initial sample under such circumstances.
Do not release product from a donor that has not had a sample tested by NAT until 180 day serology testing is performed. Initial donor sampling by NAT minimises the window period for detection of HIV, HBV, and HCV infections.
Information on the timeframe between collection and sampling should be included in the application and may be subject to GMP inspection.
The requirement to perform NAT is to significantly shorten the window period for detection of HIV, HBV and HCV infections.
Figure 1: The requirements of donor blood sample testing.
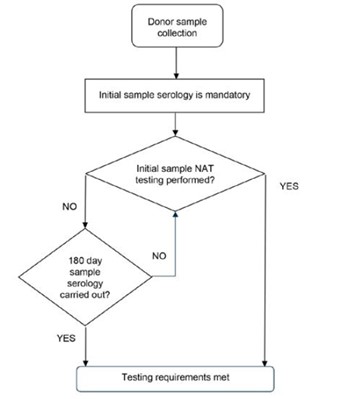
{kind=link}
Flow chart showing the process for donor blood sample testing:
- Start with 'Donor sample collection'
- Proceed to 'Initial sample serology is mandatory'
- Decision point: 'Initial sample NAT testing performed?' If Yes: Proceed directly to 'Testing requirements met' If No: Continue to next decision point
- Decision point: '180 day sample serology carried out?' If Yes: Proceed to 'Testing requirements met' If No: Return to '180 day sample serology carried out?' decision point.
The chart shows two paths to meet testing requirements: either through initial NAT testing or through 180-day sample serology if initial NAT testing is not performed.
HTLV testing may not be required of donors if a risk assessment is submitted which demonstrates that the donor, the donor’s parents, the donor’s sexual partners originate from a low risk HTLV area and that the HCT material is not a viable, leukocyte-rich cell or tissue.
Risk assessments for not performing HTLV testing should consider:
- prevalence of HTLV infection in the donor and their contacts populations; and
- the viable leukocyte content of the cells and tissues.
Prevalence sources can be from relevant scientific texts such as from the European Centre for Disease Prevention and Control: Geographical distribution of areas with a high prevalence of HTLV-1 infection. Stockholm: ECDC; 2015, or other state based sources. ‘High prevalence’ – is considered a prevalence over 1% in the general population of adults over 18 years old or prevalence of over 1/10 000 among first-time blood donors.
Examples of cells or tissue that are not considered viable, leukocyte-rich cells or tissues include, but are not limited to:
- corneas
- sclera
- skin
- heart valves
- bone
- tendons.
Sponsors should describe how manufacturing steps reduce leukocyte content from these tissues.
Subsection 11(11) – Infectious disease testing for plasma fractionation
11 Blood samples – taking and testing
Testing – living donors of HCT materials used exclusively for plasma fractionation
- The following testing must be conducted in relation to blood samples taken in accordance with paragraph (5)(a):
- serology testing for HIV-1, HIV-2, HCV, and HBsAg; and
- NAT for HIV-1, HIV-2, HBV and HCV.
Conduct serology testing for HIV-1, HIV-2, HCV, and HBsAg; and NAT for HIV-1, HIV-2, HBV and HCV on living donors of HCT materials used exclusively for plasma fractionation, who had samples taken in accordance with subsection 10(5).
Donors of HCT materials for plasma fractionation must be non-reactive to the tests stipulated. Donors of HCT materials for plasma fractionation do not require testing for syphilis and HTLV-1/HTLV-2.
Subsections 11(12) and 11(13) – Infectious disease testing for donors less than 18 months old
11 Blood samples – taking and testing Testing in relation to donors less than 18 months old
- If a donor of HCT materials is less than 18 months old, then the donor’s birth mother must be subjected to the same testing requirements that are applicable to the donor, as set out in subsections (1) to (11).
In the case of a donor of the HCT materials that are HPC(CB) only, the applicable testing requirements in this section apply only in relation to the donor’s birth mother.
Note: An infant donor of HPC(CB) only is not required to be tested in accordance with this section.
Where a donor is less than 18 months old, the birth mother must be subjected to the same sampling and testing as the infant donor as set out in subsections 11(1) to 11(11).
Where the HCT material is haematopoietic progenitor cell cord blood (HPC(CB)), the birth mother must be tested. The infant is not required to undergo testing.
The sampling and testing requirements are dependent on the state of the donor (living or deceased) and the HCT products that are being manufactured, that is, if the donor’s HCT material is being used for plasma fractionation, then the testing requirements set out under subsection 11(11) apply to the birth mother and the infant donor.
Subsection 11(14) – Infectious disease testing for donors who have consumed breast milk
11 Blood samples – taking and testing
Testing in relation to donors who have consumed breast milk
- If a donor has consumed breast milk from a person (the relevant person) within the previous six months, then the relevant person must be subjected to the same testing requirements that are applicable to the donor, as set out in subsection (1) to (11).
Where a HCT donor has consumed breast milk from a person in the previous six months, the person who provided the breast milk (the relevant person) must also undergo the same sampling and testing as the HCT donor as set out in subsection (1) to (11).
The sampling and testing requirements are dependent on the state of the HCT donor (living or deceased) and the HCT products being manufactured, that is, if the donor’s HCT material is being used for plasma fractionation, then the testing requirements set out under subsection 11(11) apply to the donor and the provider of the breast milk.
Subsections 11(15) to 11(17) – Assessment of testing results
11 Blood samples – taking and testing
Assessment of testing results
- An HCT product must be placed in quarantine, and must not be released for supply, until the results of the testing mentioned in subsections (1) to (14) are assessed.
- If the testing of a blood sample demonstrates a reactive result, then the relevant HCT product must not be released for supply.
- Subsection (16) does not apply to an HCT product that is manufactured for directed allogeneic use where the medical or dental practitioner who is responsible for the administration to, or application in the treatment of, the designated patient is notified about the testing results.
Place HCT products into quarantine while awaiting the test results for samples taken and tested in accordance with subsections 11(1) to 11(14).
No products can be released for supply until all test results are assessed and the donor/relevant persons are shown to be non-reactive/negative. Do not release the product if a blood sample is shown to be reactive to any of the tests conducted.
Where a product is for directed allogeneic use and shown to be reactive to any of the tests conducted, the product may still be released if a medical or dental practitioner is responsible for the administration/application of the product has been notified of the results.
Subsections 11(18) to 11(19) – Archiving of serum of plasma of blood samples
11 Blood samples – taking and testing
Serum or plasma of blood samples
- The serum or plasma of a blood sample taken from a donor of HCT materials in accordance with subsection (3), (4) or (6) must be:
- archived at or below minus 25°C, or in accordance with conditions that are validated or recommended by the manufacturer of the IVD medical device or in house IVD medical device used for testing the samples; and
- retained for a minimum of two years after the expiry date of the relevant HCT product, or for a period that is validated on the basis of validated data or documented evidence from relevant scientific literature.
- If:
- an HCT product has not been released for supply; and
- following the testing of a blood sample in relation to the product, a protocol or methodology for the testing changes; then, the archived serum or plasma of the blood sample must be tested in accordance with the new protocol or methodology before the product is released for supply, unless the testing is not required on the basis of a risk assessment.
Archiving serum or plasma samples
Archive serum or plasma for two years after the expiry date of the product, or for a validated period.
This requirement is in place to allow a manufacturer the ability to conduct reviews of the infectious disease status of donors or conduct additional tests if donor selection and deferral criteria change during product storage.
Failure to store a sample or loss of a sample is a 'non-conformance' and should follow internal non-conformance procedures. This may be subject to review during GMP inspection.
Retesting of archived samples
If there are changes to donor deferral during product storage (which may be for several years after the completion of processing), the archived donor blood sample may need to be re-tested based on these changes.
Before the release of the product, the suitability of the donor (based on donor deferral criteria at the time of product release) should be reviewed to ensure the donor meets all current requirements.
During the storage of the product, there may be changes to mandatory donor infectious disease testing requirements, either introduced by the manufacturer or mandated by TGA. This may include the requirement to test for additional infectious diseases in the event of a disease outbreak. In these situations, it may be required to reassess the donor before the release of product derived from their collected starting material. This must be done using archived serum or plasma, stored according to TGO 108.
The requirement to retest the donor will be based on a risk assessment conducted by the manufacturer or sponsor taking into account the nature of the risk to the recipient. TGA should be consulted if changes to donor deferral criteria will be introduced.
Section 12 - Physical assessment
Section 12 specifies the requirements for the physical assessment of donors and the minimum donor infectious disease testing requirements. Where HCT materials donated solely for the purpose for plasma fractionation to be used exclusively for export only medicines, such donors are not required to undergo physical assessment.
In addition to the medical and social history of the donor, assessment of donor blood samples and the physical assessment of the donor are further key determinants of donor acceptability.
Assessment and testing of donors are critical tiers of risk mitigation as they may be indicative of a behaviour or lifestyle of infectious disease transmission.
Subsection 12(1) - Potential donor testing for infectious diseases
12 Physical assessment;
- A physical assessment must be conducted in relation to a donor of HCT materials (other than a donor of HCT materials used exclusively for plasma fractionation in the manufacture of an export only medicine) as follows:
- in the case of a living donor (other than a living donor of human musculoskeletal tissue only) - at the time of the collection of the HCT materials; or
- in the case of a living donor of human musculoskeletal tissue only - within 30 days before or 30 days after the collection of the tissue; or
- in the case of a deceased donor - before the collection of the HCT materials.
This subsection specifies when physical assessments must be conducted. The time periods during which donor physical assessment should occur are specified to allow for the assessment to take place at a convenient time prior to surgical procedures. These are selected to be sufficiently current to allow determination of acceptability of the collection.
Subsection 11(2) - Donor physical assessments
12 Physical assessment
- A physical assessment must:
- include a clinical inspection of any physical features or characteristics of a donor (such as an abrasion, laceration, bruise, haematoma, fracture, tattoo, piercing, scar, skin lesion, or surgical incision) that may indicate that the donor poses a risk of infectious disease transmission; and
- be conducted by a person who is:
- appropriately qualified and trained;
- an employee of, or under contract with, a person engaged in the collection of HCT materials or the manufacture of HCT products; and
- demonstrate that the donor does not pose a risk of infectious disease transmission.
Determine the requirements for physical assessment of a potential donor of HCT according to a risk assessment for the tissue(s) or cells to be collected.
The exact nature of this assessment is not prescribed, each individual collection facility must determine their own assessment protocol, but it is emphasised that the physical assessment must be sufficient to ‘demonstrate that the donor does not pose a risk of infectious disease transmission’. The requirements will be influenced by the type of product and by the constraints of the clinical requirements of different patient (recipient) groups.
Document the physical assessment process. Requirements for performing and documenting the physical assessment must be specified in the dossier submitted by the sponsor to TGA for evaluation and approval. Some provisions for post-collection assessment have been made in product-specific orders.
The term ‘trained assessor’, or ‘trained interviewer’, denotes a person who has undergone training in the specific assessment and consent procedures required by the manufacturer and who has a formal relationship with the manufacturer for this purpose. The requirement for a trained assessor implies that the person undertaking the assessment has an adequate level of training and clinical skills appropriate to the assessment being undertaken, and fully understands the requirements for donor evaluation to determine suitability.
Ensure that the person performing the assessment has an appropriate level of expertise and is familiar with requirements for donor acceptance.
Document the training process in the dossier.
Schedule 1 – Ineligibility criteria for donor selection
Schedule 1 of TGO 108 describes the medical and social history that must be collected, and the potential periods of ineligibility that must be applied to a proposed donor.
Ensure you familiarise yourself with all items listed in schedule 1 and the associated requirements.
Items 1 to 5 - Known infection or potential exposure to certain viruses
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 1 | a person who is infected with:
| permanently ineligible |
| 2 | a person who is infected with:
| permanently ineligible |
| 3 | a person who has potentially been exposed to:
| ineligible until such time as the person is demonstrated not to be infected |
| 4 | a person who has potentially been exposed to:
| ineligible until such time as the person is demonstrated not to be infected |
| 5 | a person who is infected with, or has potentially been exposed to, HBV | ineligible until it has been demonstrated that the person is:
|
The ability to allow donors to be accepted after a period of ineligibility is dependent on an uninfected state of the donor being demonstrated.
For HCV, an uninfected state can be established if an individual had undergone successful anti-viral treatment, and the individual has been shown to be HCV negative by polymerase chain reaction (PCR) over a period.
For HBV, if a patient previously infected with HBV is Hepatitis B surface antibody positive (with or without core antibody positivity), but negative for both Hepatitis B surface antigen and HBV DNA, then they may be re-eligible to donate. Demonstration of exposure versus immunity to HBV must be determined by a testing algorithm that is informed from evidence in scientific literature or other reliable sources, as above. For example, donors suspected of being infected with HBV who are HBsAg negative are considered ‘immune’ if both of the following apply:
- they have an antibody titre to HBsAg at a level greater than or equal to 10 IU/L (or 10 mIU/mL)
- HBV NAT is negative.
Donors suspected of being infected with HBV, who are HBsAg negative, are considered to be ‘not exposed’ if they test negative for antibodies to HBsAg and test negative by HBV NAT.
For HBsAg negative persons who are demonstrated to be immune or never exposed, no ineligibility period applies.
| Deferral criterion | How to determine infection status | Deferral outcome |
|---|---|---|
| If known to have HBV infection | Positive HBsAg test | Ineligible |
| If suspected of having an HBV infection | Symptoms of infection or close contact with infected person established at interview but negative HBsAg test and cannot establish immunity status | Ineligible |
| If suspected of having an HBV infection | Negative HBsAg test AND Positive anti-HBc test AND Positive anti-HBs test AND Negative NAT | Immune due to natural infection previously, so no deferral required if negative NAT result confirms no current infection |
| If suspected of having an HBV infection | Negative HBsAg test And Negative anti-HBc test And Positive anti-HBs test | Immune due to hepatitis B vaccination, and so no deferral required if NAT result confirms no current infection |
Item 6 – Injected with drugs for a non-medical reason
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 6 | a person who has received an injection of any substance in connection with a use that is not a:
| ineligible for a period of at least 5 years from the last injection received by the person |
Such injections would include:
- injection of any substance in connection with a non-therapeutic use
- recreational drug use
- any procedure that is not undertaken by a registered medical or dental practitioner in Australia.
This deferral criterion is not intended to apply to individuals who have participated in medically supervised, registered clinical trials.
Item 7 - Recipients of viable, non-human cells or tissues
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 7 | a person who has been a recipient of viable animal cells or tissues | permanently ineligible |
For recipients of viable, non-human animal cells or tissue products, there is a risk of xenogeneic infections. Donors receiving such transplants are permanently ineligible.
Item 8 - At risk of prion disease
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 8 | a person who is at risk of prion disease because the person has been, or potentially been, exposed to the putative causative agent of one of the family of pathogenic transmissible spongiform encephalopathies, including:
| permanently ineligible |
Permanent deferral of a donor may need to be considered due to risk of prion disease where:
- patients have symptoms of progressive neurological disease consistent with prion disease
AND
- activities that could iatrogenically transfer prion disease have occurred.
Item 9 - Recipients of human pituitary derived hormone
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 9 | a person who has been a recipient of human pituitary-derived hormone | permanently ineligible |
Human derived pituitary hormone is currently not available in Australia. Synthetically derived products are currently used. This permanent ineligibility does not apply to proposed donors that have received synthetically derived product.
10 - At risk of acquiring a blood borne infection
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 10 | a person who has experienced any of the following events, which may give rise to a risk of acquiring a blood borne transmissible infection:
|
|
A donor with exposure to the risk of acquiring a transmissible blood borne infection such as HIV, HBV and HCV must be deferred for:
- a period that allows determination of disease development (at least 6 months from exposure)
AND
- they subsequently test negative for HIV, HBV, and HCV after this period (at least 4 months from exposure).
Item 11 - Allogeneic products
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 11 | a person who has been a recipient of allogeneic blood, blood components, human derived clotting factors, organs, cells or tissues that did not conform with this instrument | Period of ineligibility
|
If a proposed donor has received allogeneic blood, blood components, human derived clotting factors, organs, cells or tissues that did not conform with TGO 108, the sponsor must ensure that the product the donor received meets equivalent requirements to TGO 108.
This may take the form of a statement describing the international standard with which the product was compliant and confirming equivalence to TGO 108. Where any of these products is not in accordance with the requirements of TGO 108, the proposed donor is ineligible from 6 months after receiving them.
Item 12 - At increased risk due to sexual practices
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 12 | a person who has engaged in sexual activity that puts the person at an increased risk of acquiring infectious diseases that could be transmitted through blood, cells or tissues | ineligible for a period of at least 3 months from the date the person last engaged in the sexual activity |
Sexual practices that are considered to increase the risk of acquiring infectious diseases that can be transmitted through blood, cells or tissues include, but are not limited to:
- sexual activity with a sex worker
- sexual activity with someone who uses intravenous drugs
- having a partner who lives or lived in a high HIV-risk country
- or male donors, sexual activity with a male partner
- for female donors, sexual activity with a male partner who also has sex with men.
A donor meeting these criteria are ineligible for at least 3 months from the last sexual contact. The donor information that informs this deferral should be determined based on risk as relevant to the nature of the product and its use.
Item - 13 Prison inmate for 72 or more consecutive hours
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 13 | a person who has been imprisoned for a consecutive period of 72 hours or longer | ineligible for a period of 12 months from the date of release of the person from prison |
There is a known relationship between imprisonment, drug use, sexually transmitted and blood-borne virus infections such as hepatitis B and C.
Item 14 - Active infection, fever or infectious disease
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 14 | a person who has a symptomatic infection, fever or infectious illness | ineligible for a period of at least 2 weeks from the date of full recovery of the person. |
Clinical judgement should be used to determine the relevance of the infection, fever or illness to the suitability of the donor.
Determination of a disease-free state should be established before a proposed donor can be allowed to donate. This may include an algorithm and testing or specified parameters to demonstrate that an infection has cleared.
Item 15 - Travel to specific regions
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 15 | a person who has travelled to another country or region within Australia with exposure to particular epidemiological situations | ineligible for a period of time based on a risk assessment using the most up-to-date epidemiological data |
This deferral is intentionally broad to encompass unforeseen infectious disease risks such as an epidemic, pandemic, or other emerging or re-emerging infectious disease outbreak. It is the responsibility of the sponsor to demonstrate that a process is in place to monitor, assess and action epidemiological situations relevant to their products.
In the case of the COVID-19 disease pandemic, a manufacturer will need to monitor advice on the risk of infection to the safety of their product and consider how to manage the risk through deferral of donors (and potentially testing).
For travel-based deferrals, a documented procedure must be in place to record the countries where crucial infectious diseases are endemic or where there is an outbreak. The deferral may be based on a country of travel, or where an agent is endemic to a local area within that country. WHO publishes a list of countries where specific diseases such as malaria, Zika virus, Dengue fever, Ebola virus disease, West Nile Virus (WNV) are present.
Management of this requirement should be achieved through a policy for monitoring for outbreaks and notification by e-mail to TGA’s Biological Science Section (BSS) if interim measures are put in place. If there is a serious outbreak, but the risk to HCT products is considered to be minimal, you should notify TGA’s Biological Science Section (BSS) that no action is needed at this time.
Item 16 to 18 - Risk of malaria
| Item | Medical and social history criteria | Period of eligibility |
|---|---|---|
| 16 | a person who has lived in a malaria endemic region for a continuous period of 6 months or more at any time |
|
| 17 | a person (other than a person mentioned in item 16) who has visited a malaria endemic region |
|
| 18 | a person who has or has had malaria |
|
Endemic areas for malaria are published online by WHO and the Centers for Disease Control and Prevention. The definition of endemic region may be at the country level, or it may be possible to justify more specifically local areas within any one particular country.
The concept of having ‘visited’ a malaria endemic region is to be applied as consistent with WHO guidelines. In the case of malaria, visitation includes any situation where the donor has been potentially exposed to the external environment. For example, it would not include a stop-over in an airport terminal where the individual did not leave the terminal, but would include if the donor were to travel to another location (including another terminal) in a bus or private taxi. Malarial prophylaxis does not affect the malaria ineligibility periods.
TGA acknowledges that clearance of malarial parasites from HCT materials could be an alternative to this requirement. Recognising the sensitivity of the malaria parasite to gamma irradiation, HCT material subjected to irradiation with ≥ 25 kGy during manufacture of the HCT product appears sufficient to clear malaria parasites. Similarly, if the HCT product from such a donor will be terminally sterilised, the risk of malaria transmission is likely low. Thus donors who have had potential exposure to malaria may be accepted without any deferral period if the HCT material will, during manufacture, have additional treatments of either:
- a terminal sterilisation process during manufacture of the HCT product, which must be validated to ensure a maximal sterility assurance level of 10-6
- ≥ 25 kGy of gamma irradiation as an additional manufacturing step.
Item 19 - Undiagnosed febrile illness consistent with malaria
| Item | Medical or social history criteria | Period of ineligibility |
|---|---|---|
| 19 | a person who has or has had an undiagnosed febrile illness, with symptoms consistent with malaria during, or within 6 months of return from, a visit to a malaria endemic region |
|
The decision to apply this deferral criterion must be based on clinical judgement as to whether the febrile illness is consistent with malaria.
Item 20 - Active infection
| Item | Medical or social history criteria | Period of ineligibility |
|---|---|---|
| 20 | a person with an active infection that would render HCT materials collected from that person unsuitable for use in the manufacture of HCT products | ineligible until such time as the person is demonstrated no longer to be infected |
A list of infections that are relevant to the product and warrant deferral is to be established. Determination of a disease-free state includes an algorithm and at minimum the essential assays or parameters to demonstrate that an infection has cleared or will render the target cells or tissue unsuitable for manufacture.
Items 21 and 22 - Deceased donors
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 21 | a deceased person who, within 12 months prior to asystole, has been a recipient of allogeneic HCT materials or an allogeneic organ that did not conform with this instrument | permanently ineligible |
| 22 | a deceased person whose cause of death is unknown | ineligible until such time as a post-mortem examination of the person provides sufficient information to conclude that the death of the person was not caused by a transmissible disease |
If a deceased donor who, within 12 months prior to asystole, has received allogeneic HCT materials or an allogeneic organ that did not conform with TGO 108, the sponsor must ensure that the product the donor received meets equivalent requirements to TGO 108.
This may take the form of a statement describing the international standard with which the product was compliant and confirming equivalence to TGO 108. Where any of these products is not in accordance with the requirements of TGO 108, the deceased donor is permanently ineligible.
Before donation, a post-mortem is required for a deceased donor whose cause of death is unknown to ensure their death was not caused by a transmissible disease.
Items 23 to 26 - Risk of transmission of vaccination strain
| Item | Medical and social history criteria | Period of ineligibility |
|---|---|---|
| 23 | a person who has been vaccinated with a live vaccine that contains attenuated bacteria or viruses, other than a vaccine mentioned in item 24 | ineligible for 4 weeks |
| 24 | a person who has been vaccinated with a live vaccine against smallpox | ineligible for 8 weeks |
| 25 | a person who has been vaccinated with a live vaccine that contains sera of animal origin | ineligible for 12 weeks |
| 26 | a person who has been vaccinated with an unknown vaccine | ineligible for 12 months |
A proposed donor who has been vaccinated with a live vaccine, where there is a risk of transmission of the vaccine strain, must be deferred.
The length of deferral should be based on:
- knowledge of the length of persistence of the vaccine agent, or
- testing of the proposed donor, or
- based on the assessment of the risk to use of blood, cells or tissues for transplantation.
For other vaccines, manufacturers will need to consider how to manage any risk and justify any deferral.
Annex 1: Location of requirements in dossier
| Subsection | Summary of TGO 108 requirement | Relevant dossier section/s 2 | Summary of how requirement is met 3 | Referenced documents |
|---|---|---|---|---|
| 9(1)-9(3) | General requirements | 4.2. Manufacturing process | ||
| 9(4) | Blood sample testing timeframes | 4.1.2. Donor blood sampling and testing | ||
| 9(5) | Blood sample testing using IVDs | 4.1.2. Donor blood sampling and testing | ||
| 9(6) | Infectious disease testing laboratories | 4.1.2. Donor blood sampling and testing | ||
| 9(7)-(9(8) | Plasma dilution calculations | 4.1.2. Donor blood sampling and testing | ||
| 9(9) | Record keeping | 4.1.2. Donor blood sampling and testing | ||
| 9(10) | Informing donor of test results | 4.1.2. Donor blood sampling and testing | ||
| 10(1)-10(2) | Medical and social history of donors – Living donors | 4.1.1 Donor selection (Medical and Social history) | ||
| 10(3) | Medical and social history of donors – deceased donors | 4.1.1 Donor selection (Medical and Social history) | ||
| 10(4)-10(5) | Donors of HCT materials used exclusively for plasma fractionation | 4.1.1 Donor selection (Medical and Social history) | ||
| 10(6) | Donors of HCT materials that are human ocular tissue only | 4.1.1 Donor selection (Medical and Social history) | ||
| 10(7) | Change in donor history – review before release | 4.1.1 Donor selection (Medical and Social history) 4.1.3. Donor evaluation and management | ||
| 10(8)-10(9) | Review of donor history before use of collected materials | 4.1.3. Donor evaluation and management 4.2.2 Description of manufacturing process and process controls | ||
| 10(10)-10(13) | Screening requirements for donors under 18 months or who have consumed breast milk | 4.1.1 Donor selection (Medical and Social history) | ||
| 10(14) | HCT products that are manufactured for directed allogeneic use | 4.1.1 Donor selection (Medical and Social history) | ||
| 11(1)-11(2) | Blood samples for donor screening | 4.1.2. Donor blood sampling and testing | ||
| 11(3)-11(4) | Blood sampling timeframes – living donors | 4.1.2. Donor blood sampling and testing | ||
| 11(5) | Blood sampling timeframes – living donor, exclusively for plasma fractionation | 4.1.2. Donor blood sampling and testing | ||
| 11(6) | Blood sampling timeframes – deceased donors | 4.1.2. Donor blood sampling and testing | ||
| 11(7)-11(10) | Testing requirements – living and deceased donors | 4.1.2. Donor blood sampling and testing | ||
| 11(11) | Testing requirements – living donor, exclusively for plasma fractionation | 4.1.2. Donor blood sampling and testing | ||
| 11(12)-11(14) | Testing requirements - donors less than 18 months old or donors who have consumed breast milk | 4.1.2. Donor blood sampling and testing | ||
| 11(15) | Assessment of infectious disease test results – quarantine of HCT | 4.1.3. Donor evaluation and management 4.2.4 Critical steps and intermediates | ||
| 11(16) | Assessment of infectious disease test results | 4.1.3. Donor evaluation and management 4.2.4 Critical steps and intermediates | ||
| 11(17) | Assessment of infectious disease test results – directed allogeneic use | 4.1.3. Donor evaluation and management 4.2.4 Critical steps and intermediates | ||
| 11(18)-11(19) | Archiving of blood samples | 4.1.2. Donor blood sampling and testing 4.1.3. Donor evaluation and management | ||
| 12 | Physical assessment of donors | 4.1.1 Donor selection (Medical and Social history) |
Footnotes
- Therapeutic Goods Regulations 1990, Schedule 5A, Item 13.
- Suggested dossier location; actual location of information may vary depending on the nature of the product, but must be defined under this heading.
- Only a very brief summary is required, the entire dossier will be evaluated.
Page history
Title changed from 'ARGB Appendix 12 - Guidance on TGO 108: Standard for Human Cell or Tissue Products - Donor Screening Requirements' to 'Understanding donor screening rules for human cell or tissue products ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Addition of Annex 1.
Original publication.
Title changed from 'ARGB Appendix 12 - Guidance on TGO 108: Standard for Human Cell or Tissue Products - Donor Screening Requirements' to 'Understanding donor screening rules for human cell or tissue products ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Addition of Annex 1.
Original publication.