Varying biological entries on the ARTG
Purpose
This guidance is for sponsors applying for a variation to the Australian Register of Therapeutic Goods (ARTG) entry of a biological.
Note that an ARTG entry is not limited to the information visible in the public ARTG entry. It also includes any supporting information provided with the dossier or subsequent variations that are held by TGA and were considered to be relevant to the initial decision.
This guidance is:
- for biologicals regulated under the Biologicals Regulatory Framework that are included in the ARTG under Part 3-2A of the Therapeutic Goods Act 1989
- not for biological medicines (which are prescription medicines and registered under Part 3-2 of the Therapeutic Goods Act 1989)
For matters that would have been relevant to the initial decision to include the biological in the ARTG:
- You need to request and receive our approval for any variations or changes to or in relation to the biological.
- This is to satisfy a condition imposed at the time of inclusion in the ARTG.
Wait for our approval
Do not implement a variation before we have approved it, otherwise you risk breaching the provisions of the Therapeutic Goods Act 1989:
- You are breaching a condition of inclusion in the ARTG if you implement a variation before the Secretary has approved it.
- Penalties may apply (sections 32EF (criminal offences) and 32EG (civil penalties) of the Therapeutic Goods Act 1989).
This restriction does not apply to notifications, where the proposed change may be introduced at the time of submitting the variation application to the TGA.
When to use this guidance
Use this guidance to determine if your change meets any of the following categories:
- corrects an ARTG entry
- creates a separate and distinct good
- is a safety-related variation
- has the potential to impact quality, safety or efficacy
If your change does not fit within one of these categories then it is likely that, under the legislation, you are not varying your entry in the ARTG.
If you are unsure whether you need to make an application, email us at bloodandtissues@health.gov.au
Steps
For a sponsor to determine if a change is a variation to an ARTG entry and how to apply for approval, the following processes are involved:
-
1. Determine if the change would have been relevant to the inclusion decision
-
2. Determine if the change is a correction to your ARTG entry
-
3. Determine the variation category
-
4. Determine the supporting document requirements
-
5. Submit your application
-
6. Prepare and submit your cover letter and supporting documents
-
7. Pay your application fee
-
8. TGA screening of your application
-
9. Pay your evaluation fee
-
10. TGA evaluation of your supporting documents
-
11. Making a decision on your application
-
12. Implement the change(s) if approved
Determine if the change would have been relevant to the inclusion decision (step 1)
The questions below may help identify those changes ‘that would have been relevant to the decision to include the biological in the ARTG’.
These questions are not exhaustive and are the only indicators as to whether the change would have been relevant to the decision.
Does the change:
- Impact on the current process or justification that demonstrates conformity to a standard? Relevant standards may include:
- product-specific standards (TGO 108)
- the labelling standard (TGO 107)
- the donor screening and infectious disease requirements (TGO 108)
- pharmacopoeial monographs (default standards on Pharmacopoeias).
- Alter a critical in-process control or release specification?
- Have the potential to alter the justification used to support a critical in-process control or release specification?
- Justification can be by reference to limits set down in a standard, international guidance, process validation or from published literature.
- When information becomes available that alters the appropriateness of the justification, this could impact on the initial decision to accept the specification.
- Nullify or invalidate previous validation studies?
- A manufacturing process or method needs to be supported by current validation studies submitted and accepted by TGA.
- Re-validation should occur and data be submitted if the changed process or method is not supported by current studies.
- Alter a manufacturing process or test method, including changes to infectious disease test kits?
- Alter the key quality and safety parameters for a critical material?
- Critical materials are defined as supplies or reagents that come in direct contact with the cell or tissue during any stage of manufacture. For example:
- chemically defined buffers, transfer pipettes, and primary containers or collection kits
- containers and materials of human and animal origin
- Key parameters are those that determine suitability for the intended purpose e.g. quality control specifications, sterility and biocompatibility.
- Critical materials are defined as supplies or reagents that come in direct contact with the cell or tissue during any stage of manufacture. For example:
- Introduce a new manufacturing site or change the scope of manufacturing steps performed at a current manufacturing site, which relate to that product?
- Generate a new Master Cell Bank or Working Cell Bank?
- Alter the formulation or composition of the finished product?
- Alter the approved risk-benefit profile?
- Alter the intended clinical use/therapeutic indication (including safety-related changes)?
If the answer to any of these questions is, ‘Yes’, it is likely that the variation will have the potential to affect the quality, safety or efficacy of the product and will require submission to TGA and approval prior to implementation. For changes that require submission to TGA go to Step 3 of this guidance to determine the category of the change and if it requires evaluation or is ‘self-assessable’.
If the answer to all of these questions is, ‘No’, then it is possible that an application to vary your ARTG entry is not required. The exception is if the change could be considered a correction to your ARTG entry (Step 2, below).
Note
Your change could still impact compliance with Good Manufacturing Practice (GMP) requirements.
Epidemiological situations
It is a requirement of compliance with TGO 108 (or TGO 105) that epidemiological situations are monitored and that appropriate action is taken where quality or safety of the product could be impacted. TGA approves the process in place at facilities to perform this monitoring, review and potential action in response to epidemiological situations.
As part of this approval process it is a requirement that TGA is notified where an interim donor deferral or manufacturing measure is put in place. Email this notification to bloodandtissues@health.gov.au. Note that this is not considered a change that would have been relevant to the inclusion decision, so is not considered a variation to your entry.
However, the following two situations would usually be considered to be variations to an ARTG entry:
- where there is a change to the TGA-approved process for management of epidemiological situations
- where an interim measure is assessed as needing to be implemented on a more permanent basis. In this case, we consider the issue to be no longer simply management of an epidemiological situation, because the situation is now permanent and requires a change to your donor selection criteria to achieve conformity to TGO 109 section 9 (or TGO 105 section 12(8) for FMT products).
Determine if the change is a correction to your ARTG entry (step 2)
If you want to correct an ARTG entry that is incomplete or incorrect, you will need to apply to the TGA under subsection 9D(1) of the Therapeutic Goods Act 1989.
Note that an ARTG entry is not limited to the information visible in the Register. It also includes any supporting information provided with the dossier or subsequent variations that are held by TGA and were considered to be relevant to the initial decision.
Correction to an ARTG entry is not applicable to export only biologicals. If you are varying the ARTG entry of your export only biological, you must submit a notification (see step 3).
Corrections
Apply for a correction when:
- There are spelling or grammatical errors in the information visible in the ARTG entry.
- The information visible in the ARTG entry is incorrect or absent.
- There are typographic errors in the Product Information or labels that need to be corrected to align with the ARTG entry details.
- You identify that a substantial error or omission of information was made in the information submitted previously in support of the inclusion of the biological. This does not include editorial and other future updates to documents.
Changes that are not corrections
The change will not be considered a correction to the ARTG details under subsection 9D(1):
- If editorial changes are made to information previously submitted in support of the inclusion of the biological. This is not a change to your ARTG entry. For example, corrections to a policy, standard operating procedure or validation report that do not change the intention or outcome.
- If the change is relevant to the decision to include your biological in the ARTG or any later decision to vary the ARTG entry. These are changes to your entry that require approval under 9D(3), as the change has the potential to impact on the quality, safety or efficacy of the biological.
If your change would not have been relevant to the initial decision (Step 1), and is not a correction to your ARTG entry (Step 2), then it is likely that no application to vary your entry in the ARTG is required.
When you are correcting an ARTG entry
There is no fee to correct an ARTG entry for a biological. To complete your online application, you need a fee exemption code:
- Draft an application (Step 5. Start your application).
- Contact the Biologicals Team at bloodandtissues@health.gov.au requesting a ‘fee exemption code’ for an application under subsection 9D(1) to correct an ARTG entry, making reference to the draft application number.
- Once a fee exemption code has been generated, the application form can be submitted.
- Prepare and submit a cover letter to support the change (Step 6. Prepare your cover letter).
Determine the variation category (step 3)
The variation category depends on three steps:
- Determine if the change creates a separate and distinct good
- Determine if the change is safety-related
- Other changes with the potential to impact quality, safety or efficacy.
Determine if the change creates a separate and distinct good
If you are making a change that creates a separate and distinct good (see regulation 11A, Therapeutic Goods Regulations 1990), you are not varying the entry for an existing biological, but creating a new one.
Class 1 and 2 biologicals
For Class 1 and 2 biologicals, changing any of the following will create a separate and distinct good:
- applicable standards (e.g. TGO 107-109, default standards on Pharmacopoeias)
- if there is a new standard that becomes applicable, then the good becomes a separate and distinct good
- if the standard is updated, then any change made to the biological to ensure conformity to the updated standard is a variation
- intended clinical use
- principal manufacturer.
Class 3 and 4 biologicals
For Class 3 and 4 biologicals, changing any of the following will create a separate and distinct good:
- product name
- dosage form
- formulation or composition
- therapeutic indication
- type of container, regardless of container size
- principal manufacturer.
Export only biologicals
For export only biologicals, changing any of the following will create a separate and distinct good:
- active ingredient
- dosage form
- principal manufacturer.
New biologicals based on parent (already approved) biologicals
If you are creating a separate and distinct good:
- Apply for a new biological based on a parent biological
- Do not apply for a variation.
Determine if the change is safety-related
Safety-related variations made under section 9D(3AA) of the Therapeutic Goods Act 1989 are limited to variations that:
- reduce the patient population
- for example, limiting the use of the biological
- add a warning or precaution
- for example, about an adverse event or interaction.
If the therapeutic indication changes as a result of a safety-related variation, it will not be treated as a separate and distinct good.
This section is not applicable to export only biologicals. If you are varying the ARTG entry of your export only biological, you must submit a notification (see under: Other changes with the potential to impact quality, safety or efficacy).
If you change the therapeutic indication of a class 3 or 4 biological, but not as a result of a safety-related variation, you are creating a separate and distinct good:
- Apply for a new biological based on a parent biological
- Do not apply for a variation under section 9D(3AA).
Justify
Explain how your variation meets the criteria for a safety-related variation.
Safety-related variations always require changes to the release documentation outlining warning statements e.g. Product Information. Where appropriate to support the changes, submit these as additional supporting documents.
If the proposed change is more than adding a simple word or phrase to release documentation or is intended to reflect new quantitative findings from a clinical trial or other type of study, the TGA is likely to need to evaluate supporting data.
However, it is possible that safety-related changes can be considered ‘self-assessable’ and do not require evaluation of the supporting data. These may be identified on TGA screening of the application. The evaluation fee does not apply to self-assessable variations.
You can read more about safety-related requests for prescription medicines, including those that may not require evaluation of the supporting data are available, in form 9D(92) Safety-related request to vary an ARTG entry [DOCX 140 KB].
Assessing your justification
We will grant your request if we agree that your variation:
- meets the criteria for a safety-related variation; and
- does not create a separate and distinct good.
We may need to evaluate the supporting documents to make this decision.
Sometimes we contact you
We contact you when we become aware of safety-related information that requires a safety-related variation to one of your ARTG entries. This might be because:
- we have identified a signal during post-market monitoring; or
- other ARTG entries of a similar kind have been varied for safety-related reasons.
In this instance a response will be sought from you, which may result in the need to submit a safety-related variation.
Changes that are not safety-related
If your variation is not considered to be safety-related, it may still have the potential to impact quality, safety or efficacy.
Other changes with the potential to impact quality, safety or efficacy
This section refers to changes that have the potential to affect the quality, safety or efficacy of the biological (under section 9D (3A) and 9D (3AC)), provided that the change does not create a separate and distinct good.
Based on the level of review required, variations under 9D (3A) and 9D (3AC) are split into four application categories:
- notifications
- self-assessable variations
- minor variations
- major variations
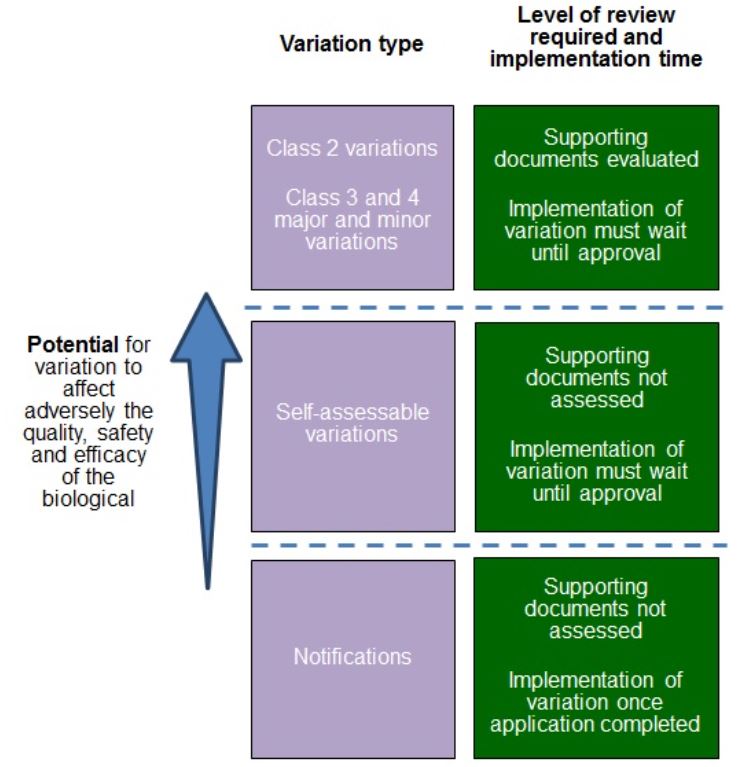
{kind=link}
This diagram illustrates different levels of variation types and their corresponding review requirements. The diagram is organized in three rows with two columns each, and includes an upward-pointing blue arrow on the left indicating increasing "Potential for variation to affect adversely the quality, safety and efficacy of the biological."
From top to bottom, the rows are:
- First row (highest level):
- Left column (purple): "Class 2 variations, Class 3 and 4 major and minor variations"
- Right column (green): "Supporting documents evaluated; Implementation of variation must wait until approval"
- Middle row:
- Left column (purple): "Self-assessable variations"
- Right column (green): "Supporting documents not assessed; Implementation of variation must wait until approval"
- Bottom row (lowest level):
- Left column (purple): "Notifications"
- Right column (green): "Supporting documents not assessed; Implementation of variation once application completed"
The rows are separated by dashed lines, and the text in each cell is displayed in white against either purple (left column) or dark green (right column) backgrounds.
The definitions for the main application categories of variations are provided below.
| Application category | Definition |
|---|---|
| Notifications | Applies to specific variations made under section 9D (3AC) of the TG Act that have been determined to pose a very low risk when the defined conditions are met. These variations do not require evaluation of information and would be expected to have minimal effect on the quality, safety or efficacy of the biological. The changes can be implemented once the application (form, fee and supporting documents) have been submitted to the TGA. |
| Self-assessable variations | Applies to variations made under section 9D (3A) of the TG Act that do not require evaluation of information and would be expected to have minimal effect on the quality, safety or efficacy of the biological. The changes must be approved by the TGA before implementation. |
| Minor variation | Applies to a variation made under section 9D (3A) of the TG Act that requires the evaluation of quality and manufacturing information, other than a change that is a major variation for the biological or that would result in the biological becoming separate and distinct from other biologicals. The changes must be approved by the TGA before implementation. |
| Major variation | Applies to a change to the entry for any of the following:
The changes must be approved by the TGA before implementation. |
The following sections provide guidance on the types of changes that do not require evaluation of the supporting documents (notifications, self-assessable variations). By default, if the change does not meet the definition of a notification or self-assessable variation then the change will be treated as a minor or major variation, as defined above.
Notifications
Notifications are types of variations that have been determined to pose a very low risk. The TGA has concluded that their implementation would not affect the established quality, safety and efficacy of an included biological. Notifications still require an application to the TGA, but do not require evaluation.
Variations can be submitted as a notification if:
- your variation is one of the notifications detailed in the tables below and
- all conditions that are relevant to that notification are met.
As TGA experience with variations to ARTG entries for biologicals increases, we anticipate adding to the list of notifications.
Tables of notifications
| Change | Notification code | Conditions |
|---|---|---|
| Changes as a result of amendments to an applicable standard (e.g. pharmacopoeial or TGO) | PT |
|
| Change | Notification code | Conditions |
|---|---|---|
| Changes to the donor selection criteria, including the medical and social history questionnaire | DS |
|
| Changes to infectious disease test kits | TK |
|
| Critical material change | SM |
|
* Note that changes to some critical materials may have a more significant impact on the product than others and may require evaluation of the supporting data. For example, a change to the quality of a growth supplement (critical material) can have a significant effect on the culture conditions and would often require re-validation of the manufacturing process; a change to a primary container (critical material) may require re-validation of product stability.
| Change | Notification code | Conditions |
|---|---|---|
| Changes to in-process specifications | MI |
|
| Critical material change | SM |
|
| Removal of a product listed in the entry for a Class 2 biological | BR | You must submit details of the change |
* Note that changes to some critical materials may have a more significant impact on the product than others and may require evaluation of the supporting data. For example, a change to the quality of a growth supplement (critical material) can have a significant effect on the culture conditions and would often require re-validation of the manufacturing process; a change to a primary container (critical material) may require re-validation of product stability.
| Change | Notification code | Conditions |
|---|---|---|
| Addition of a new site performing secondary packaging or secondary storage | MA |
|
| Changes to site performing testing (including quality control and infectious disease testing) | MT |
|
| Removal of a site of manufacturer | MR |
|
| Change | Notification code | Conditions |
|---|---|---|
| Changes to release specifications | BS |
|
| Reduction in shelf-life of finished product or shipping timeframes | BT |
|
| Change | Notification code | Conditions |
|---|---|---|
| Changes to the product labels or supporting documents (e.g. Product Information, Consumer Medicine Information and patient card) | LC | The change must be one of the following:
|
Export only biologicals changes
| Change | Notification code | Conditions |
|---|---|---|
| Changes to the information in the ARTG entry of export only biologicals. | EX |
|
Self-assessable variations
For some variations with the potential to impact the quality, safety or efficacy of the biological (including some safety-related variations), the supporting documents may not require evaluation.
Applications that do not require evaluation of supporting documents are termed ‘self-assessable’ variations.
To determine if your variation is self-assessable:
- check the defined self-assessable variations or
- assess the risk
- Self-assessable variations are not applicable to export only biologicals. If you are varying the ARTG entry of your export only biological, you must submit a notification.
Defined self-assessable variations
Your supporting documents do not require evaluation (i.e. are self-assessable) if:
- your variation is one of the defined self-assessable variations and
- all conditions that are relevant to that variation are met.
As TGA experience with variations to ARTG entries for biologicals increases, we anticipate adding to the list of defined self-assessable variations.
Tables of defined self-assessable variations
| Change | Conditions |
|---|---|
| Addition of a new working cell bank |
|
| Change | Conditions |
|---|---|
| Variation was previously approved for another biological |
|
Assessing risk
If your change is not listed in one of the tables of defined self-assessable variations, then you will need to assess the risk of the change adversely affecting the quality, safety or efficacy of the biological. You will need to submit this risk assessment with your variation request.
The variation must have only a low potential to adversely affect the quality of the biological in order to not require evaluation of the data by the TGA.
Note that if a subsequent review of the change identifies a greater potential to impact the quality safety or efficacy of the biological then it may require evaluation of the supporting data.
Contact us
If you are unsure on the application category or whether your supporting documents require evaluation, contact the Biologicals Team at bloodandtissues@health.gov.au.
Determine the supporting document requirements (step 4)
Most variations require documents to be provided to support an application. Corrections to ARTG entries under subsection 9D(1) of the Therapeutic Goods Act 1989 may not require any supporting documentation.
The level of supporting documentation required for variations depends on:
- the complexity of the change; and
- the potential to adversely affect the quality, safety or efficacy of the good.
The following supporting information and documents should be provided, where applicable:
- details as to how the variation changes information held by TGA in regard to a biological on the ARTG
- documents to support the variation e.g. validation data, operating procedures, literature references, updated specifications.
Supporting documents may include copies of:
- amended procedural documentation and specifications (including a marked up and a clean copy to allow side-by-side comparison)
- amended labels and Product Information sheets
- new test methods
- method or process validation studies
- published literature used to support a change
- safety and/or efficacy study reports (complete reports)
- any other relevant information.
The guidance provided in Step 3 (Determine the variation category) will help you determine whether the supporting documents require evaluation or not.
Submit your application (step 5)
To make your application:
- Know your product/s ARTG number
- Decide how many applications to make
- Fill out the form
Know your product/s ARTG number
You need to know the ARTG number for the biological. Once you enter this number in the form, the data in the current ARTG entry will be placed into the form:
- you will be able to change information in most of the fields
- some fields are locked, because they cannot be changed in a variation application.
Decide how many applications to make
Consider the following:
- multiple variations in a single application
- multiple products
- multiple ARTG entries
Multiple variations in a single application
You can combine multiple variations to a biological or biologicals in a single ARTG entry within one application (except for safety-related changes), where they fit more than one category of variation.
The following categories of variations can and cannot be combined in a single application:
- can combine: correcting an entry, notifications, self-assessable and minor variations
- cannot combine: major variation or safety-related changes.
Consider the following aspects carefully when deciding whether to submit a single application with multiple variations or make a separate application for each variation:
- Where a single application contains multiple variations in relation to biological in a single ARTG entry, the TGA will consider the acceptability of each of the variations individually.
- Self-assessable variations submitted in a single application with one or more variations that require evaluation cannot be approved and implemented until a decision is made for all variations in the application.
- As the TGA considers each variation on its merits, the non-approval of any of the variations within an application containing multiple changes would not result in rejection of the entire application.
- Where the TGA identifies questions with any of the variations, the decision on the application will be delayed until all issues have been resolved.
Multiple products
For class 2 biologicals, you can apply to vary some or all products contained within a single ARTG entry. This does not apply to class 3 and 4 biologicals as there is only a single product included under each ARTG entry.
Multiple ARTG entries
One application must be made for each ARTG entry.
Where you are introducing the same variation to separate entries on the ARTG, an application is required for each ARTG entry, but we may evaluate them at the same time.
More than one application may be submitted together, as a single ‘submission’, where all of the applications selected have the same:
- class of biological
- sponsor billing address
- principal manufacturer undertaking the release for supply step
- product standards, for Class 1 or Class 2
- active ingredient, for Class 3 or Class 4.
If the applications do not meet these criteria then they must be submitted separately. However, if the applications are from the same sponsor they may still be treated as abridged applications if:
- part of the dossier supporting the applications is sufficiently similar for their evaluation to be abridged
- your cover letters for the applications refer to each other and you request that the applications be considered together
- you explain how the applications are similar.
Reduction or waiver of the evaluation fees may apply to simultaneous submissions and abridged applications.
Note that at this time there can only be one application under review for a specific ARTG entry at a time.
Fill out the form
For all variations to a biological included in the ARTG, you must submit an application via TGA Business Services. For information on accessing and completing the form, see the Biologicals application form - a step-by-step guide.
Prepare and submit your cover letter and supporting documents (step 6)
This step applies to requests for all variations.
Prepare a cover letter for:
- corrections
- safety-related variations
- other variations (notifications)
- other variations (self-assessable)
- other variations (not self-assessable)
- variations that impact multiple applications
Follow our general dossier requirements for all submitted supporting information.
Corrections cover letter
In your cover letter for a correction to an ARTG entry, include:
- the variation category and section of the Therapeutic Goods Act 1989 that you are making your application under:
- 'This application is to correct an ARTG entry of a biological and is made under subsection 9D(1) of the Therapeutic Goods Act 1989'
- reference to the application number (following submission of the online application form; Step 5)
- an explanation of why this variation is simply a correction i.e. what information is incomplete or incorrect and how
- the proposed change/s, such as a marked up copy of a document.
If you refer to previously submitted documents, provide sufficient details for us to be able to locate the documents easily.
Safety-related cover letter
In your cover letter for a safety-related variation, include:
- the variation category and section of the Therapeutic Goods Act 1989 that you are making your application under:
- 'This application is a safety-related variation for a biological and is made under subsection 9D(3AA) of the Therapeutic Goods Act 1989'
- reference to the application number (following submission of the online application form; Step 5)
- an explanation of how the proposed change meets the criteria for a safety-related variation
- the variations, such as a marked up copy of a document
- if you are including supporting documents, provide a list of these.
Other variations (notification) cover letter
In your cover letter for a notification, include:
- the variation category and section of the Therapeutic Goods Act 1989 that you are making your application under:
- 'This application is a notification for a variation to a biological with potential to impact the quality, safety or efficacy (but not resulting in the reduction of quality, safety or efficacy) and is made under subsection 9D(3AC) of the Therapeutic Goods Act 1989'
- reference to the application number (following submission of the online application form; Step 5)
- details of the variations including the notification code for each variation
- a declaration that your changes meet the conditions that apply to the notification code
- the proposed timing and approach to implementing the variation, including the expected time period during which the pre-variation and post-variation products might be supplied concurrently
- a list of the supporting documents being provided.
If you refer to previously submitted documents, provide sufficient details for us to be able to locate the documents easily.
If third party documents are provided directly to TGA to support the change, you also need to provide a letter from the relevant third party stating that you have the authority to rely on them as supporting information.
Other variations (self-assessable) cover letter
In your cover letter for a self-assessable variation, include:
- the variation category and section of the Therapeutic Goods Act 1989 that you are making your application under:
- 'This application is a self-assessable variation for a variation to a biological with potential to impact the quality, safety or efficacy (but not resulting in the reduction of quality, safety or efficacy) and is made under subsection 9D(3A) [or subsection 9D(3AA) for safety-related changes] of the Therapeutic Goods Act 1989'
- reference to the application number (following submission of the online application form; Step 5)
details of the variations - a justification for why you think the variation is self-assessable, with sufficient detail for us to be able to determine quickly whether it is appropriately classified
- a declaration that your changes meet the conditions that apply to defined self-assessable variations, if applicable
- if the variation is not a defined self-assessable variation, an explanation of how your risk assessment supports a low-risk classification
- the proposed timing and approach to implementing the variation, including the expected time period during which the pre-variation and post-variation products might be supplied concurrently
- a list of the supporting documents being provided.
If you refer to previously submitted documents, provide sufficient details for us to be able to locate the documents easily.
If third party documents are provided directly to TGA to support the change, you also need to provide a letter from the relevant third party stating that you have the authority to rely on them as supporting information.
Other variations (not self-assessable) cover letter
In your cover letter for a variation with potential to impact the quality, safety or efficacy (but not so as to reduce the safety, quality or efficacy) when evaluation of supporting documents is required, include:
- the variation category and section of the Therapeutic Goods Act 1989 that you are making your application under:
- 'This application is for a [variation/minor variation/major variation] to a class [2/3/4] biological with potential to impact the quality, safety or efficacy and is made under subsection 9D(3A) of the Therapeutic Goods Act 1989'
- reference to the application number (following submission of the online application form; Step 5)
- details of the variations, and a justification as to why the quality, safety or efficacy has not been reduced
- the proposed timing and approach to implementing the changes, including the expected time period during which the pre-variation and post-variation products might be supplied concurrently
- a list of the supporting documents provided.
If you refer to previously submitted documents, provide sufficient details for us to be able to locate the documents easily.
If third party documents are provided directly to TGA to support the change, you also need to provide a letter from the relevant third party stating that you have the authority to rely on them as supporting information.
Variations that affect multiple products
If you are making similar applications to vary biologicals in multiple ARTG entries (e.g. simultaneous and abridged applications):
- include the application numbers from TGA Business Services of the other applications; and
- explain how the applications are similar, and indicate whether a reduction or waiver of the evaluation fee is being sought.
Determine the supporting document requirements
Most variations require documents to be provided to support an application.
The level of supporting documentation required for variations depends on:
- the complexity of the change; and
- the potential to impact adversely on the quality, safety and efficacy of the good.
The following supporting documents should be provided, where applicable:
- where the variation changes information held by TGA in regard to a biological on the Register; and
- documents to support the variation e.g. validation data, operating procedures, literature references, updated specifications
Corrections to ARTG entries may not require any supporting documentation.
Potential supporting documents may include copies of:
- amended procedural documentation and specifications (including a marked up and a clean copy to allow side-by-side comparison)
- amended labels and Product Information sheets
- new test methods
- method or process validation studies
- published literature used to support a change
- safety and/or efficacy study reports (complete reports)
- any other relevant information.
Pay your application fee (step 7)
There is no application or evaluation fee for making corrections to an ARTG entry for a biological under subsection 9D(1). The use of the fee exemption code in the application form prevents invoicing for the application fee.
For all other applications, you need to pay the application fee at the time the application is submitted. Following submission, the acknowledgement of successful submission invites you to print a copy of your invoice.
For information on payment methods go to:
We will not consider your application until you have paid your application fee.
For details of the current fees, go to:
Screening of your application (step 8)
Once we receive your application, application fee and supporting data (if applicable), we review your application and decide:
- if the appropriate variation category has been applied
- whether the supporting documents require evaluation
- the appropriate evaluation fee.
Screening is not performed on notifications. Once the application, fee and supporting data have been received they will be processed (Step 11).
Note that where we do not agree with the variation category you have assigned for a change to be a self-assessable request, we will inform you that the change does require evaluation and provide you with a justification.
Letter
At the completion of the screening we will send you a letter informing you of:
- the variation category
- the decision, for:
- corrections to ARTG entries [9D(1) applications]
- self-assessable variations, when unaccompanied by other variations (Go to Step 12).
- for all other applications:
- whether we accept the application for evaluation (sufficient supporting documents are provided)
- the evaluation fee (invoicing will follow receipt of the letter)
- the anticipated TGA timeframe for evaluation and decision.
Screening timeframe
We aim to accomplish the screening of most applications in 15 working days.
Our legislated timeframe is 30 days for notification of whether we will evaluate the application (regulation 16GB, Therapeutic Goods Regulations 1990).
No timeframe is set for corrections to ARTG entries [applications under section 9D(1)].
Evaluation fees
An additional fee applies to the application if the supporting documents require evaluation.
The fee depends on the variation categories as outlined below:
- safety-related
- potential to impact quality, safety or efficacy
- variation to class 2 biological
- minor variation
- major variation.
Standard evaluation fees are defined in the Schedule of fees and charges.
However, we are also able to reduce or waive the evaluation fee, so that it corresponds to the level of supporting documents and assessment required [regulations 45(2) & 45(4) of the Therapeutic Goods Regulations 1990. We might reduce the fee:
- if you make another application(s) in relation to therapeutic goods at the same time and the following circumstances apply (simultaneous submission):
- the goods to which each application relates contain the same active ingredient
- the information given in support of each application has sufficient commonality, in respect of the goods, that the goods may conveniently be simultaneously evaluated
- if the Secretary has information relating to the goods that enables the evaluation procedure to be abridged (abridged application).
Pay your evaluation fee (part 9)
Following screening of your application, if evaluation of the supporting information is required you will be sent an invoice for an evaluation fee.
There is no evaluation fee for:
- making changes corrections to the ARTG entry under section 9D(1)
- self-assessable variations.
For details of the current fees, go to:
For information on payment methods go to:
Evaluation of your supporting documents (step 10)
We will evaluate most applications within the TGA target timeframes below. The letter we send you at the end of the screening will state the anticipated TGA timeframe for evaluating your application.
Screening and evaluation timeframes for variations to biologicals
| Application category | TGA target timeframes (screening + evaluation) |
|---|---|
| Correction to an ARTG entry | 15 working days in total |
| Self-assessable variation | 15 working days |
| Safety-related changes | TGA processes as soon as possible |
| Variations of class 2 biologicals | 15 + 45 working days |
| Minor variation | 15 + 45 working days |
| Major variation | 15 + 255 working days |
When you have applied for multiple variations that fit more than one category, the applicable timeframes will reflect the highest level of category change. For example, if you have applied for one or more self-assessable variation and one minor variation, the timeframes will be those of the minor variation.
The legislated evaluation time for variation applications under sections 9D(3A) and 9D(3AA) is 255 working days (regulation 16GD, Therapeutic Goods Regulations 1990). There are some exceptions to the target internal timeframes, for example:
- priority will be given to safety-related changes
- when an application with a target timeframe of 45 days is complex, we will take longer than 45 working days for the evaluation and decision
- you will be advised of a longer timeframe for evaluation of your application in the letter we send to you following screening.
Requests for information
We may ask you for more information under section 32JA of the Therapeutic Goods Act 1989.
It is an offence not to comply with such a notice or to provide information that is false or misleading in a material particular.
We must allow you at least 14 days from when you receive the notice, to provide the information and documents. The response period will be clearly stipulated in the section 32JA letter. If we have not given you sufficient time to respond, you may ask us for an extension in writing by contacting the Biologicals Team at bloodandtissues@health.gov.au.
The period of time from when we send a request under section 32JA to when you respond, does not count towards TGA working days.
Making a decision on your application (step 11)
or notifications, the approval letter will be sent out to you within a few working days of receiving your application and supporting data and the fee being paid.
For other variations, once we have made our decision, we will send you a decision letter. We make our decisions on all the applications within a simultaneous submission at the same time.
Read the decision letter carefully, especially the list of changes approved and the timeframe for implementation. If you have applied for more than one variation in an application, it is possible that we may approve some of the variations and not others.
Approval with conditions on the inclusion
As part of the approval the Secretary may consider whether additional conditions need to be imposed under section 32EE of the Act.
Rejection
If the decision is to reject some or all of the variations in your application, the decision letter will include both:
- a statement of the reasons for the decision; and
- information on your rights to seek a review of the decision.
Implement the change (step 12)
Wait for approval before implementing a change. You are breaching a condition of inclusion in the ARTG if you implement a variation before the Secretary has approved it.
If a variation results in improved quality or safety, we may also impose a new condition of inclusion under section 32EE with a timeframe for implementing the change.
On rare occasions, some changes beyond your control may take place before the TGA can approve them (for example, a change to a test kit used by a contracted facility). In these cases, it is your responsibility to submit a request to the TGA as soon as you become aware of the change.
Page history
Inclusion of export only biologicals.
Minor updates.
Clarification provided for Multiple ARTG entries (Step 5)
Link to new Biologicals application form guidance (Step 5).
Addition of notifications.
Original publication.
Published as part of Consultation: Guidance on variations to biologicals included in the Register.
Inclusion of export only biologicals.
Minor updates.
Clarification provided for Multiple ARTG entries (Step 5)
Link to new Biologicals application form guidance (Step 5).
Addition of notifications.
Original publication.
Published as part of Consultation: Guidance on variations to biologicals included in the Register.