Changing or transferring conformity assessment certificates
Guidance for notifying the TGA about substantial changes to, or transfers of, conformity assessment certificates.
Purpose
The purpose of this guidance is twofold:
- To assist manufacturers in assessing whether proposed changes to a conformity assessment certificate amount to 'substantial changes' and if so, what the manufacturers' obligations are for the purposes of meeting the automatic conditions on a conformity assessment certificate and the requirements of the conformity assessment procedures.
- To set out the events that trigger the transfer of a conformity assessment certificate and its associated responsibilities to a new entity (i.e. a new manufacturer).
This is a guide only. Sponsors and manufacturers are encouraged to seek independent professional advice to ensure they comply with their legislative and regulatory requirements under the therapeutic goods legislation. This document will evolve over time. Updates and clarifications will be included as required. Feedback on the guidance is always welcome.
This guidance is for manufacturers that have been issued conformity assessment certification by the Therapeutic Goods Administration (TGA) and are planning to make a change. This guidance also includes events that trigger a transfer of a conformity assessment certificate, and its associated responsibilities, to a new entity.
Compliance with conformity assessment procedures and ‘substantial changes’ condition on TGA certificates
A manufacturer holding a TGA-issued conformity assessment certificate is subject to a number of automatic conditions imposed under Section 41EJ of the Therapeutic Goods Act 1989 (the Act) and is required to ensure ongoing compliance with the applicable conformity assessment procedures.
One of the conditions imposed is that the manufacturer (or person in respect of whom the certificate is issued1) and having regard to the type of certificate issued, will notify in writing the Secretary of the Department of Health, of any plan for substantial changes to:
- quality management systems
- the product range covered by those systems
- the product design of kinds of medical devices.
This means that any planned changes that are relevant to the certificates that a manufacturer holds, must be notified to the TGA, who will review and approve these changes on behalf of the Secretary.
Similarly, the conformity assessment procedures variously require that if after assessment, a manufacturer plans to make any substantial changes to the quality management system, to the kinds of device to which a system has been applied, or to the design or intended purpose of any devices which have undergone design or type examination, then they must notify the Secretary and arrange for assessment of that change.
The purpose of this guidance is to assist a person in respect of whom a conformity assessment (CA) certificate has been issued, to determine whether their proposed changes require notification to the TGA in accordance with the legislation. This guidance also clarifies that notification (and application) of a planned substantial change ensures that, under the Act, the manufacturer remains compliant with the conditions imposed on the certificate.
For a planned substantial change to come into effect, it is necessary for the manufacturer to make an application for a conformity assessment certificate as set out in section 41EB of the Act.
Notification and application of a planned substantial change, in itself, does not result in the conformity assessment certificate being changed. The notification will be reviewed before agreement that the changed device continues to comply with requirements of its CA certification and may also result in an updated version of the existing certificate being issued with regards to the substantially changed medical device.
Urgent changes that are vital to public health and safety should be implemented immediately
Labelling changes that add a contraindication, warning or precaution vital to public health and safety should be implemented immediately, with relevant change documentation maintained under the manufacturer’s QMS. These types of changes - including the notification to the TGA - should be carried out in accordance with the Uniform Recall Procedure for Therapeutic Goods (URPTG).
However, the manufacturer is also required to notify the TGA of the change as part of their application for recertification. Documentation supporting any change must be made available to the TGA at any time, and upon request.
What is considered to be a substantial change
A substantial change is a change to the manufacturer’s QMS or medical devices, including IVD medical devices, that are expected to impact the quality, safety or performance of the devices.
The concept of a substantial change is linked to the principles of safety and performance in the context of a risk-based regulatory framework to control the risk of medical devices, including IVD medical devices.
Examples of what the TGA considers to be “substantial changes”, and “not substantial changes” are provided in Section 4 and Section 5 respectively of this guidance.
How to notify the TGA of a substantial change
Conformity assessment certificates are issued to the manufacturer, and it is their responsibility to notify the TGA. The manufacturer, or an applicant who is authorised to perform this task on their behalf, notifies the TGA by submitting a “Substantial change notification and application” conformity assessment application through their TGA Business Services (TBS) portal. This serves the dual purpose of evidencing the manufacturer’s compliance with their notification obligation under the Act and meeting the requirement to arrange for an assessment of that change, by lodging an application for a new version of conformity assessment certificate for approval to implement the proposed change.
Nomenclature update
The TGA conformity assessment application forms have been updated. What was previously known as an application for ‘change to an existing certificate’ is now known as ‘substantial change notification and application’. The change in naming aligns with recognition that the notification of a change is processed via submission of an application. There is no separate notification process required or expected
Method for notification and application for a substantial change
Substantial changes must be notified and assessed by the TGA prior to supplying any products (kinds of devices) affected by those changes.
A substantial change notification and application is submitted via the TGA Business Services (TBS) portal. For guidance on how to do this, please go to: Application for conformity assessment certificates. The application is subject to an initial application fee.
Note
On the first page of the TBS CA application form, under ‘Applicant Type Details’, ensure you select the option for “substantial change notification and application”
In the Applicant’s reference field, ensure you include "Change to conformity assessment certificate".
What happens after submission?
Manufacturers should allow adequate lead-time for the TGA to complete the assessment of the proposed changes prior to implementation. The assessment may require an on-site audit, which will be incorporated into the TGA’s audit programme. In some circumstances, and subject to the nature of the change and extent of documentary evidence available to support the change management process undertaken by the manufacturer, the TGA may review the documentation and approve the change, prior to carrying out a surveillance audit.
Following receipt of an effective application2 the TGA will determine the level of assessment necessary to verify that the device will continue to comply with the applicable provisions of the essential principles through the appropriate conformity assessment procedures and issue an updated version of the existing conformity assessment certificate if the supporting evidence is considered acceptable (see Flowchart 1).
Note
The term “substantial changes” is not defined under the Act. The TGA therefore provides extensive guidance on what it considers to be a substantial change. Examples and flowcharts are set out at Section 4 and Appendix A of this guidance.
However, it is not possible to specify what would be considered a substantial change for each of the vast range of devices on the market.
Manufacturers must have change control procedures in place that consider the impacts of the change and whether the change is “substantial” and requires notification and application to the TGA to comply with the automatic conditions of the TGA conformity assessment certificate.
Process for notification of a substantial change
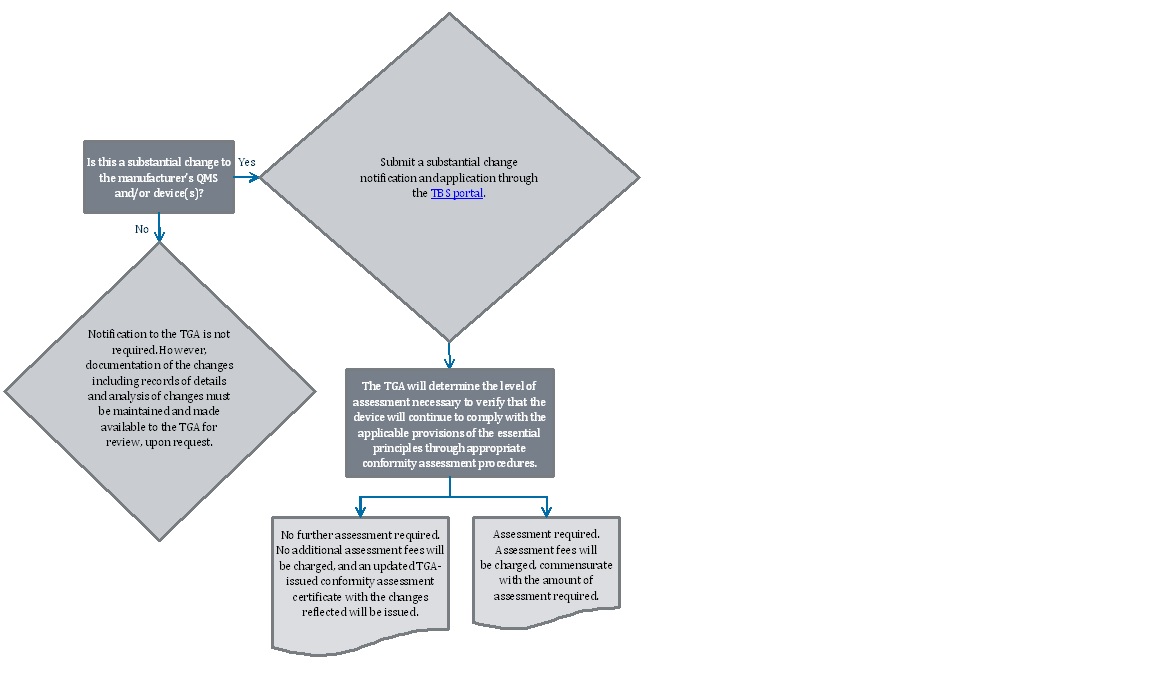
{kind=link}
The image shows a flowchart describing the process for handling changes to a manufacturer's Quality Management System (QMS) and/or device(s). The flowchart consists of decision points, actions, and outcomes.
- The flowchart starts with a decision point: "Is this a substantial change to the manufacturer's QMS and/or device(s)?"
- If the answer is "Yes":
- Action: "Submit a substantial change notification and application through the TBS portal."
- Next step: "The TGA will determine the level of assessment necessary to verify that the device will continue to comply with the applicable provisions of the essential principles through appropriate conformity assessment procedures."
- This leads to two possible outcomes: a. "No further assessment required. No additional assessment fees will be charged, and an updated TGA-issued conformity assessment certificate with the changes reflected will be issued." b. "Assessment required. Assessment fees will be charged, commensurate with the amount of assessment required."
- If the answer is "No":
- Outcome: "Notification to the TGA is not required. However, documentation of the changes including records of details and analysis of changes must be maintained and made available to the TGA for review, upon request."
The flowchart uses a combination of rectangular, diamond, and rounded rectangle shapes to represent different elements of the process. Arrows connect the shapes to show the flow of decision-making and actions.
Following review of a substantial change notification and application, the TGA will consider updating the details of any affected entries in the Australian Register of Therapeutic Goods (ARTG) on its own initiative. If a decision to vary the entry or entries is made, the sponsor will be notified when the update is complete. In some cases, for example when there are a large number of affected ARTG entries, the sponsor may be requested to submit a Device Change Request (or variation) application, and the corresponding application fees will apply.
For further details relevant to the variation of ARTG entries for medical devices refer to the TGA website: Essential Principles – consent for noncompliance (if applicable).
Tools to determine substantial changes
This section presents general principles and flowcharts to assist manufacturers to determine whether a change is considered substantial, and therefore requires notification to the TGA. Several examples of changes that are commonly made to medical devices, including IVD medical devices, are included in the Appendices, however these examples are not an exhaustive list and should be considered as representative only.
Changes to quality management systems
This section applies to changes to the manufacturer’s QMS, including changes to manufacturing facilities or suppliers included on the TGA-issued conformity assessment certificate; or their relevant scopes that are outlined on the certificate; or changes to validated processes that are performed at any of these sites. Flowchart 2 and Flowchart 3 within this guidance, detail questions to assist applicants and manufacturers in determining if a change to the manufacturer’s QMS is considered to be substantial, and therefore requires notification to the TGA. Specific examples are provided in Appendix A.
Changes to a manufacturing facility and/or relevant scope
For example, a change in:
- name and/or address of the manufacturer
- scope of existing manufacturing facilities, including manufacturing steps
- addition or removal of a manufacturing facility along with associated activities.
Sometimes a name change may be the result of a certificate transfer due to the disposal of a business, its amalgamation with another manufacturer, the death or bankruptcy of the owner, or the winding up of a business. For these situations, please refer to the section Transfers of conformity assessment certificates.
Changes to manufacturing process
This includes any changes to the controls applied under a QMS for validated processes, in particular, changes to a process where validation is required to mitigate the risks relating to that process, or to ensure that the requirements that are essential for the safe and proper use of the device can be met. This is particularly important where the risks, if unmitigated, may adversely impact patients or users. For example, a change to:
- a drug coating process for a medical device
- the methods used for the verification of purchased products if those products are starting materials for a medical device that is intended to incorporate a medicinal substance or a material of animal origin
- a packaging process that may impact the sterile integrity of the medical device
- a manufacturing process for a medical device that might impact pre-sterilisation bioburden
- a sterilisation method for a medical device (e.g. ethylene oxide (EO) to gamma, gamma to e-beam, etc.)
- the replacement (e.g. as a result of decommissioning) or addition of sterilisation equipment for a medical device
- a viral inactivation process related to a medical device, including an IVD medical device
- the manufacturing quality control procedures for a medical device, including an IVD medical device (e.g. parametric release for sterilisation process, aeration or dwelling period for EO sterilisation, substantial change to batch release testing)
- the methods or controls for determining shelf life for a medical device, including an IVD medical device
- a manufacturing process affecting the concentration and composition (i.e. formulation) of reagents for an IVD medical device, which may impact the active components in the assay, and hence the device performance and stability.
Changes to a supplier identified on a TGA-issued conformity assessment certificate and/or relevant scope on the certificate
For example, a change in:
- name and/or address of a supplier identified on the certificate
- scope of activities for a supplier already identified on the certificate including any manufacturing process or activity that is outsourced by the supplier (e.g. abattoir) that may impact the specification of the material/component provided by the supplier
- addition of a new supplier to the certificate
- removal of a supplier already identified on the certificate.
Changes to the type of conformity assessment procedure
Each TGA-issued conformity assessment certificate identifies the conformity assessment procedures applied by the manufacturer. Any subsequent changes to the conformity assessment procedure applied by the manufacturer, for example, changing from a Schedule 3, Part 4 (Production Quality Assurance Procedures) system to a Schedule 3, Part 1 (Full Quality Assurance Procedures) system, will require submitting a new initial application of a CA certificate.
Changes to the kinds of medical devices, including IVD medical devices
A change to the kinds of devices to which the QMS has been applied must be notified to the TGA. Subsection 41EJ (3) of the Act, and the conformity assessment procedures in Schedule 3 of Therapeutic Goods (Medical Devices) Regulations 2002 (the Regulations), set out the various circumstances that a manufacturer is required to notify such changes. The kinds of devices covered within the scope of a QMS certificate is specified on the certificate under Device Categories.
For IVD medical devices, the requirement to notify the TGA of changes to the individual products under each ‘kind of device’ (i.e. the device category as listed on the Schedule 3, Part 1 conformity assessment certificate) is based on classification of the device and possible risks associated with the change (with regard to safety and performance). In general, a system-based approach that relies on the manufacturer’s QMS to identify and control the changes made to Class 2 and 3 IVD medical devices included within the scope of a TGA-issued conformity assessment certificate is possible.
This approach will only be accepted if the affected IVDs remain ‘the same kind of device’, and there is no major change to performance claims, methodology or processes used during manufacture, the environment the device is intended to be used in, or to the intended user group. Documentation of the changes including records of the details and analysis of changes made to Class 2 and 3 IVD medical devices should be made available to the TGA for review, upon request.
The Regulations specify that references to kinds of medical devices, in the context of conformity assessment procedures including changes, also include a reference to an individual medical device.
Flowchart 2: Changes to the manufacturer’s QMS
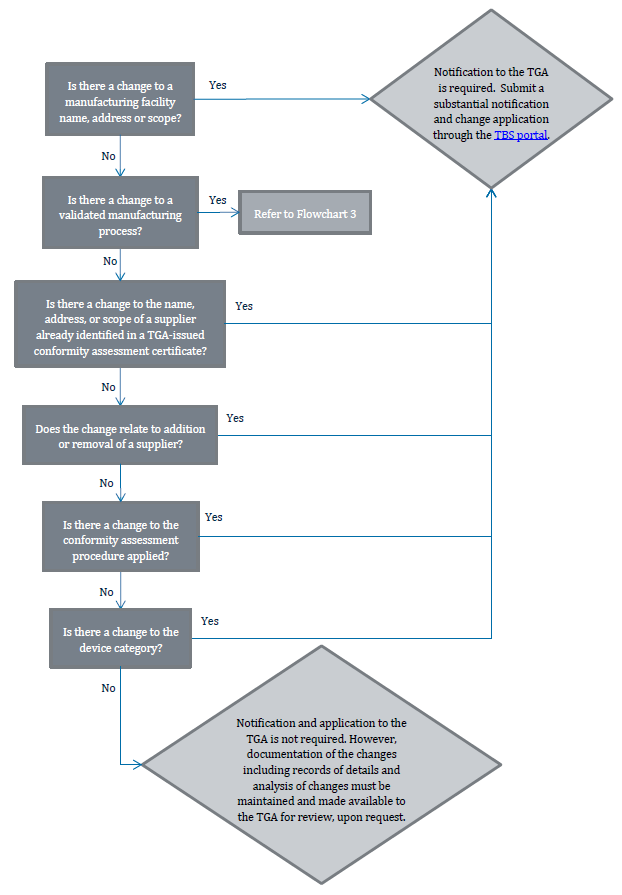
{kind=link}
The image shows a flowchart detailing the process for determining whether notification to the TGA (Therapeutic Goods Administration) is required for various changes related to manufacturing and suppliers. The flowchart consists of decision points (questions) and outcomes, connected by arrows indicating the flow of decision-making.
The flowchart progresses as follows:
"Is there a change to a manufacturing facility name, address or scope?"
If Yes: "Notification to the TGA is required. Submit a substantial notification and change application through the TBS portal."
If No: Proceed to next question
"Is there a change to a validated manufacturing process?"
If Yes: "Refer to Flowchart 3"
If No: Proceed to next question
"Is there a change to the name, address, or scope of a supplier already identified in a TGA-issued conformity assessment certificate?"
If Yes: Leads to the same outcome as the first question
If No: Proceed to next question
"Does the change relate to addition or removal of a supplier?"
If Yes: Leads to the same outcome as the first question
If No: Proceed to next question
"Is there a change to the conformity assessment procedure applied?"
If Yes: Leads to the same outcome as the first question
If No: Proceed to next question
"Is there a change to the device category?"
If Yes: Leads to the same outcome as the first question
If No: Proceed to final outcome
Final outcome if all answers are No:
"Notification and application to the TGA is not required. However, documentation of the changes including records of details and analysis of changes must be maintained and made available to the TGA for review, upon request."
The flowchart uses rectangular boxes for questions, diamond shapes for main outcomes, and arrows to show the flow between decisions. The TBS portal mentioned is highlighted in blue, indicating it may be a hyperlink in a digital version of this flowchart.
Flowchart 3: Changes to manufacturing processes
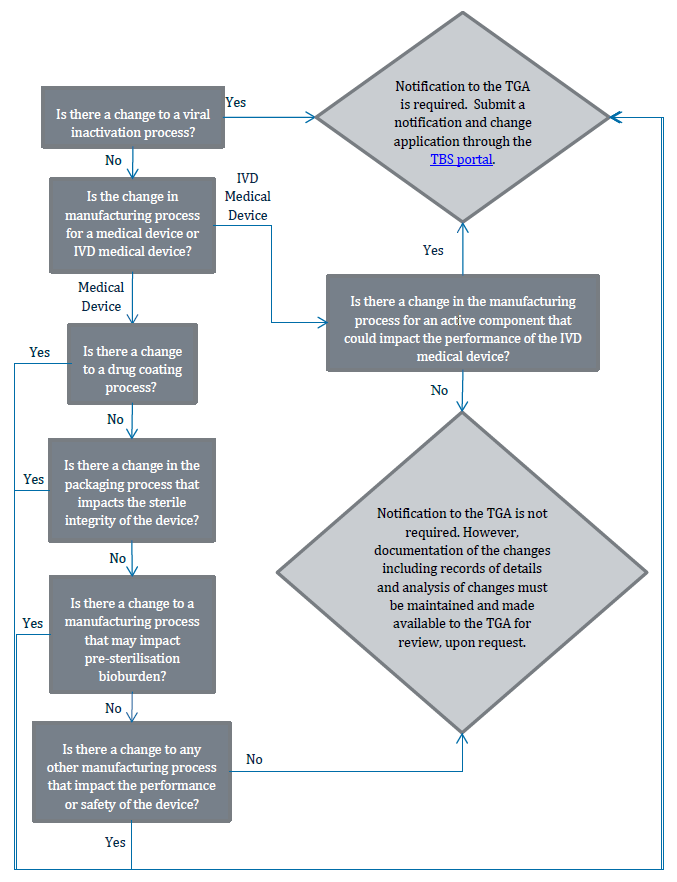
{kind=link}
The image shows a flowchart detailing the process for determining whether notification to the TGA (Therapeutic Goods Administration) is required for various changes in medical device manufacturing processes. The flowchart consists of decision points (questions) and outcomes, connected by arrows indicating the flow of decision-making.
The flowchart progresses as follows:
1. "Is there a change to a viral inactivation process?"
- If Yes: "Notification to the TGA is required. Submit a notification and change application through the TBS portal."
- If No: Proceed to next question
2. "Is the change in manufacturing process for a medical device or IVD medical device?"
- If IVD Medical Device: "Is there a change in the manufacturing process for an active component that could impact the performance of the IVD medical device?"
- If Yes: Leads to TGA notification requirement
- If No: "Notification to the TGA is not required. However, documentation of the changes including records of details and analysis of changes must be maintained and made available to the TGA for review, upon request."
- If Medical Device: Proceed to next question
3. "Is there a change to a drug coating process?"
- If Yes: Leads to TGA notification requirement
- If No: Proceed to next question
4. "Is there a change in the packaging process that impacts the sterile integrity of the device?"
- If Yes: Leads to TGA notification requirement
- If No: Proceed to next question
5. "Is there a change to a manufacturing process that may impact pre-sterilisation bioburden?"
- If Yes: Leads to TGA notification requirement
- If No: Proceed to next question
6. "Is there a change to a manufacturing process that impact the performance or safety of the device?"
- If Yes: Leads to TGA notification requirement
- If No: No TGA notification required, but documentation must be maintained
The flowchart uses rectangular boxes for questions, diamond shapes for main outcomes, and arrows to show the flow between decisions. The TBS portal mentioned is highlighted in blue, indicating it may be a hyperlink in a digital version of this flowchart.
Changes to product design
This section applies to all medical devices with TGA-issued conformity assessment certification, including IVD medical devices, and not just those kinds of devices that are subject to design or type examination procedures. A modification to a device may involve changes to its design, functionality, variants, materials, packaging, specifications and storage conditions, that are likely to introduce new hazards, alter the likelihood, severity, or detectability of harm, or consequences that were not previously documented by the manufacturer in their risk analysis and therefore may impact compliance with the essential principles.
Flowcharts 4 and 5 within this guidance detail specific questions and answers to assist applicants and manufacturers in determining if a change to product is considered to be substantial and requires notification to the TGA and submission of a “substantial change notification and application.”
Changes to final product design specifications
The following examples require a substantial change notification and application to be submitted. These include changes to:
- magnetic resonance (MR) status of a device (i.e. change from MR unsafe to MR conditional)
- addition of new variants outside the range previously approved by the TGA
- mechanical, electrical, physical or biological properties of the finished device
- the indications for use of the device, including patient populations, anatomical locations, intended user or environment of use; and in addition, for IVD medical devices – extension of recommended specimen types, or changes to stability of specimen types (e.g. environmental conditions, duration of storage, or living donor versus cadaveric specimens)
- the analytical and clinical performance characteristics for an IVD medical device as a result of a change in reagent formulation
- an IVD medical device for self-testing that may increase the risk of error in the use of the device, handling of the sample, or interpretation of results, or that may increase the complexity of use of the device for the user.
For the purpose of this document, the TGA considers “intended purpose” as being interchangeable with “intended use” and “indications for use”.
Changes in materials for medical devices (excluding IVD medical devices)
For example, changes to:
- the materials or formulations used in the medical device, particularly when they may impact the biological safety or the mechanical performance of the finished product
- the species, origin, or source of animal or microbial origin materials in medical devices, or the viral inactivation process on material that is used as a component of a medical device
- the manufacturing process, quantity or quality of medicinal substance incorporated in a medical device
- materials used during manufacturing that may result in new processing residues/degradants that impact the safety and performance of the finished product
- packaging that may impact the sterile integrity of the medical device.
Changes to the supplier listed on the TGA CA certificate or manufacturing methods of the raw materials are considered changes to the manufacturer’s QMS. These changes may impact the safety and performance of final products. Substantial change notification and application to the TGA is required.
Changes in materials for IVD medical devices
For Class 4 IVD medical devices, changes in materials for IVD medical devices require a substantial change notification and application to be submitted to the TGA. The extent of assessment or review by the TGA will depend on the materials changed and the possible impact on the performance characteristics of the IVD medical device. For example, a change to a generic reagent (e.g. buffer) requires notification, but is unlikely to require further assessment. However, for changes to active components such as capture antibodies, it is expected that extensive validation would be conducted by the manufacturer. The TGA would review the manufacturer’s validation data to ensure that the device remains compliant with the essential principles.
Changes to reagent formulation, such as component concentration, are considered as changes to design specifications (see sub-section about Changes to design specifications).
For Class 1-3 IVD medical devices that are within the scope of the manufacturer’s TGA-issued QMS certificate, provided there is no change to the active components in the formulation for these lower class IVDs, a substantial change notification and application is not required to be submitted to the TGA.
Changes in the supplier of reagent components identified on the conformity assessment certificate are considered changes to the manufacturer’s QMS.
Changes in storage, shelf-life, and packaging for medical devices, including IVD medical devices
Changes that may impact the sterile barrier integrity of a device, or its performance following storage are considered substantial, and therefore require notification to the TGA. For example, changes in:
- primary packaging materials or configuration (e.g. from tray in tray, to pouch in pouch)
- storage or transport conditions
- shelf-life/stability claims, unless the medical devices, including IVD medical devices, are low risk devices and there are no changes to the previously accepted stability validation protocol or the assigned acceptance criteria.
Flowchart 4: Changes to product design
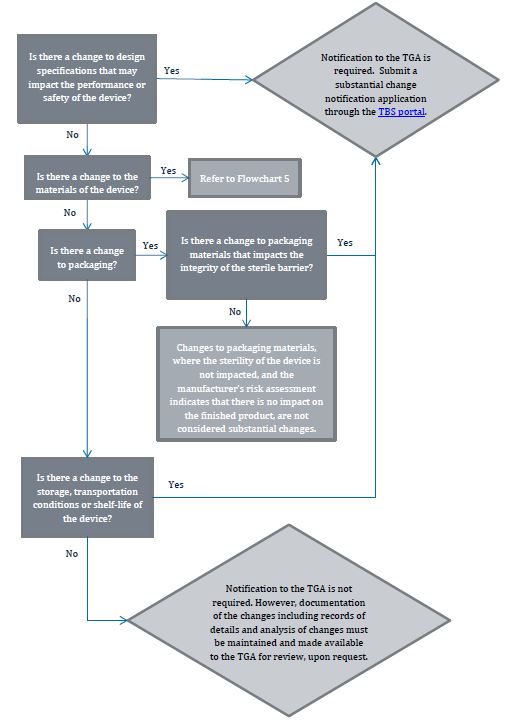
{kind=link}
The image shows a flowchart detailing the process for determining whether notification to the TGA (Therapeutic Goods Administration) is required for various changes to medical devices. The flowchart consists of decision points (questions) and outcomes, connected by arrows indicating the flow of decision-making.
The flowchart progresses as follows:
1. "Is there a change to design specifications that may impact the performance or safety of the device?"
- If Yes: "Notification to the TGA is required. Submit a substantial change notification application through the TBS portal."
- If No: Proceed to next question
2. "Is there a change to the materials of the device?"
- If Yes: "Refer to Flowchart 5"
- If No: Proceed to next question
3. "Is there a change to packaging?"
- If Yes: "Is there a change to packaging materials that impacts the integrity of the sterile barrier?"
- If Yes: Leads to TGA notification requirement
- If No: "Changes to packaging materials, where the sterility of the device is not impacted, and the manufacturer's risk assessment indicates that there is no impact on the finished product, are not considered substantial changes."
- If No: Proceed to next question
4. "Is there a change to the storage, transportation conditions or shelf-life of the device?"
- If Yes: Leads to TGA notification requirement
- If No: "Notification to the TGA is not required. However, documentation of the changes including records of details and analysis of changes must be maintained and made available to the TGA for review, upon request."
The flowchart uses rectangular boxes for questions and statements, diamond shapes for main outcomes, and arrows to show the flow between decisions. The TBS portal mentioned is highlighted in blue, indicating it may be a hyperlink in a digital version of this flowchart.
Flowchart 5: Changes to product materials
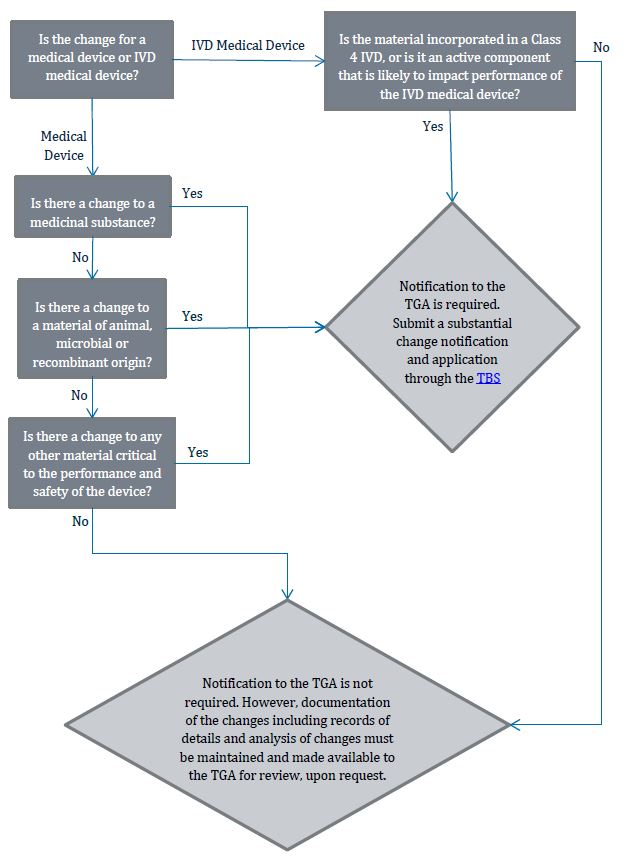
{kind=link}
The image shows a flowchart detailing the decision process for determining if notification to the TGA (Therapeutic Goods Administration) is required for changes to medical devices or IVD (In Vitro Diagnostic) medical devices.
The flowchart begins with the question: "Is the change for a medical device or IVD medical device?"
If it's an IVD Medical Device, the next question is: "Is the material incorporated in a Class 4 IVD, or is it an active component that is likely to impact performance of the IVD medical device?" If the answer is "No," the process moves to the final stage where notification is not required.
If it's a Medical Device, the flowchart proceeds through a series of questions:
1. "Is there a change to a medicinal substance?"
2. If No: "Is there a change to a material of animal, microbial or recombinant origin?"
3. If No: "Is there a change to any other material critical to the performance and safety of the device?"
If the answer is "Yes" to any of these questions, or if the answer was "Yes" to the IVD question, the outcome is: "Notification to the TGA is required. Submit a substantial change notification and application through the TBS portal."
If the answer is "No" to all these questions for a Medical Device, or "No" to the IVD question, the outcome is: "Notification to the TGA is not required. However, documentation of the changes including records of details and analysis of changes must be maintained and made available to the TGA for review, upon request."
This flowchart provides guidance on when changes to medical devices or IVDs require TGA notification and when they don't, emphasizing the importance of documentation in all cases.
Changes to the information to be provided with medical devices, including IVD medical devices
According to the Regulations, the intended purpose of a kind of medical device is ascertained from one or more of the following:
- information provided with the device (labelling)
- instructions for Use (IFU)
- patient Information Cards and Patient Information Leaflets (PIC/PILs)
- any advertising materials relating to the device
- technical documentation describing the mechanism of action of the device.
In addition, essential principle 13 of the Regulations requires some specific content to be included in the information provided with a device. This includes, among other things, the:
- intended purpose and intended patient group for the device
- conditions under which the device should be stored
- warnings in relation to the use of the device.
Therefore, some changes to labelling and the IFU may be considered substantial changes requiring notification to the TGA. Examples include (but are not limited to):
- changing information regarding the safe use of the device
- a labelling change from single use to reusable
- adding a ‘non-pyrogenic’ claim on the labelling of a device
- adding a new procedure for a use or purpose not originally assessed
- changing the MR compatibility status of a device
- changing the storage conditions that are considered critical for the performance of the device
- changing the name of the device
- changes to the IFU for extension or amendment of indications including use in a different patient population, or via a different surgical procedure
- amendment of instructions for use of the device.
For changes to the layout/typographical/administrative/grammatical content of the IFU, the manufacturer is not required to submit a substantial change notification and application to the TGA. However, records containing details and analysis of these changes, including version control should be maintained and made available to the TGA for review, upon request.
For changes that reduce indications or restrict patient populations, the requirement to notify the TGA is dependent on the potential risks associated with the change. The reasons for the change and consideration to the impact that the change will have on the overall risk-benefit profile of the device.
Flowchart 6 details specific questions and answers to assist applicants and manufacturers in determining if a change to information to be provided with a medical device, including an IVD medical device, is considered to be substantial and therefore requires submission of a substantial change notification and application to the TGA prior to implementation.
The IFU and labelling of a device can be an important part of mitigating the residual risk associated with the use of the device. If this information is changed on the existing labelling and IFU, for example to add or remove a contraindication, warning, or other important information about safe use of the device, then actions in accordance with the URPTG may be required for devices already in the field.
Flowchart 6: Changes to information to be provided with a medical device, including an IVD medical device

{kind=link}
The image presents a flowchart outlining the decision process for determining whether notification to the TGA (Therapeutic Goods Administration) is required for changes to a medical device. The flowchart consists of three decision boxes and two outcome boxes.
The decision process follows these steps:
1. "Is there a change to the intended purpose of the device?"
- If Yes, proceed directly to the notification required outcome.
- If No, move to the next question.
2. "Is there any change relevant to contraindications, warnings, adverse events or other critical information regarding safety?"
- If Yes, proceed to the notification required outcome.
- If No, move to the next question.
3. "Are there changes to the information relevant to storage and transport conditions or shelf-life of the device?"
- If Yes, proceed to the notification required outcome.
- If No, proceed to the notification not required outcome.
The two possible outcomes are:
1. "Notification to the TGA is required. Submit a substantial change notification and application through the TBS portal." (TBS is hyperlinked, likely to the relevant submission portal)
2. "Notification to the TGA is not required. However, documentation of the changes including records of details and analysis of changes must be maintained and made available to the TGA for review, upon request."
This flowchart guides users through assessing whether changes to a medical device require formal TGA notification or simply internal documentation.
What is not considered a substantial change
The following examples illustrate some types of changes that would not be considered by the TGA as substantial, and therefore do not require notification and application:
- changes to non-critical parts or materials or suppliers that do not affect the final product’s acceptance criteria established by the manufacturer
- changes to manufacturing equipment or processes which have negligible impacts on the final product’s safety and performance or are unlikely to increase the level of risk imposed to the user or patient
- changes to the artwork, colour, font, or layout of the packaging and labelling of a device that does not alter compliance with essential principle 13
- changes to manufacturing processes by suppliers identified on the conformity assessment certificate, including the processes performed by suppliers to those identified suppliers, unless the changes:
- require validation/re-validation
- result in a change to the specifications of materials/components or final product
- result in a change to the process parameters
- result in a broadening of the specifications or acceptance criteria applied to in-process or product release testing.
Although notification to the TGA is not required for the above changes, the Regulations require the manufacturer to ensure they continue to meet the requirements of their QMS for the handling of the change.
The manufacturer must maintain documentation of any changes as part of the QMS for traceability and recordkeeping purposes. Documentation of the changes must be available for review by the TGA upon request.
Examples of changes that do not require an application for a change to an existing certificate to be submitted to the TGA are provided in Appendix B.
Transfers of conformity assessment certificates
Transfers of a TGA-issued conformity assessment certificate to a new person or body corporate as a result of certain events are required to be notified to the TGA within 3 months following the transfer, with the relevant supporting evidence.
You must notify the TGA of any planned changes in relation to a TGA-issued conformity assessment certificate transfer. Ensure you notify the TGA in accordance with this guideline and implement the proposed substantial change(s) after the TGA’s approval. Such changes often result in issuance of new certificates and require a new Australian Register of Therapeutic Goods (ARTG) entry.
Events that trigger a transfer
Division 4.3 of the Regulations applies to a manufacturer of a medical device in respect of whom a conformity assessment certificate is issued and includes provisions about when a conformity assessment certificate is taken to be transferred to a new person or to a body corporate.
The Regulations state that such transfers occur where the manufacturer:
- dies, becomes bankrupt, or is wound up
- disposes of the business or amalgamates with another manufacturer
- changes its name.
The manufacturer ceases to exist, becomes bankrupt, or is wound up
Regulation 4.6 of the Regulations provides that in this case:
- the personal legal representative, trustee in bankruptcy or liquidator is taken to be the person in respect of whom the certificate is issued (i.e. the new holder) and
- the new holder must notify the TGA of that event within 3 months of it occurring; and
- provide the TGA with sufficient documentary evidence of the relevant event (Regulation 4.10).
Disposal of business or amalgamation with another manufacturer
Regulation 4.7 of the Regulations provides that where the name of the manufacturer changes because either:
- the manufacturer agrees to dispose of a business concerned with the manufacture of the medical device and it is agreed that the disposal is to include the transfer of the relevant conformity assessment certificate
- the manufacturer, being a body corporate, amalgamates with another body corporate under a different name
Then:
- the person to whom the business is disposed of, or body corporate with whom the manufacturer amalgamates is taken to be the person in respect of whom the certificate is issued (i.e. the new holder) and
- the new holder must apply for the name of the manufacturer to be changed on the conformity assessment certificate within 3 months of it occurring and
- provide the TGA with sufficient documentary evidence of the relevant event (Regulation 4.10).
Manufacturer changes its name
Regulation 4.8 of the Regulations states that if the name of the manufacturer is changed, the manufacturer, as renamed, is taken to be the person in respect to whom the certificate is issued. The manufacturer, as renamed must:
- notify the TGA within 3 months after the name is changed, and
- provide the TGA with sufficient documentary evidence of the relevant event (Regulation 4.10).
Documentary evidence may include proof of manufacturer name (such as Australian Business Number (ABN) or Registered business name) and updated labelling for the device(s) reflecting the new manufacturer name.
When there are organisational changes or a transfer to a new legal entity (partially or wholly), this is not considered a manufacturer name change. This change will require transfer of the conformity assessment certificate(s) or issuance of new certificate(s).
Examples of events that result in transfers that require notification to the TGA are provided in Appendix C.
When does the transfer take place
The TGA does not make a determination as to when the transfer actually takes place. This is determined by the timing of the relevant event (e.g. the bankruptcy) as set out in Division 4.3 of the Regulations.
Once an event occurs, the new person or business is taken to be responsible for meeting the requirements related to the conformity assessment certificate, regardless of whether the TGA has:
- been notified of the transfer or change, or
- issued a new version of the conformity assessment certificate.
If a conformity assessment certificate is transferred because of the operation of Regulation 4.6, 4.7, or 4.8:
- the legal and regulatory responsibilities transfer to the new holder of the certificate; and
- the new holder can continue to produce the medical device(s) covered by the certificate while the certificate remains valid.
Manufacturers (and prospective new manufacturers) should seek their own advice about:
- the impact of Regulations 4.6, 4.7 and 4.8 on their status and
- their obligations under the Act and its associated regulations when a conformity assessment certificate is transferred.
If the TGA becomes aware that an event that triggers a certificate transfer has not been notified within 3 months of it occurring, the TGA may suspend or revoke the conformity assessment certificate related to the event.
Method for notification and application for a certificate transfer
A notification and application to transfer a conformity assessment certificate should be lodged by the new or ‘receiving’ entity via the TBS portal - external site. For guidance on how to do this, please go to Application for conformity assessment certificates.
Regulation 4.10 states that if a person is required to notify the TGA of an event that results in transfer of conformity assessment certificates, the person must also provide the TGA with sufficient documentary evidence of the relevant event (e.g. evidence of amalgamation of a business, evidence of the bankruptcy of a manufacturer, etc).
Transfers must be notified to the TGA by the ‘receiving’ entity within 3 months of the change.
On the first page of the TBS CA application form, under ‘Application Type Details’, ensure you select the option for ‘Initial application’
In the Applicant’s reference field, ensure you include the Transfer of conformity assessment certificate
Complete and include the Transfer of conformity assessment certificates form as part of your supporting evidence.
Appendix A: Examples of substantial changes
It is not possible to provide examples that represent every type of substantial change that may occur for the wide range of medical devices on the market. Manufacturers must have change control processes in place that consider the impact of proposed changes on the safety and performance of the device, and whether the change requires notification to the TGA.
The following examples are not all-inclusive and may not be applicable in all cases3.
Quality management system – Medical devices, including IVD medical devices
- The manufacturer intends to change the source of raw material used in a medical device, from porcine origin to bovine origin. This may impact the quality, performance or specifications of the finished medical device. Data to support the appropriate verification and validation of processes for viral inactivation or minimisation needs to be assessed. This is considered a substantial change.
- The manufacturer of a tissue heart valve decides to add an alternate abattoir for the supply of bovine pericardium to avoid any potential shortages of the current supplier, with no changes to the existing viral inactivation processes that are undertaken by the manufacturer. This is considered a substantial change. Evidence of the manufacturer’s supplier control for the addition of the new abattoir, verification of bioburden results for the tissue sourced from the new abattoir, along with risk management review is required to be provided for assessment by the TGA.
- The manufacturer of a medical device changes the sterilisation process cycle parameters such as process temperature or load size, with no changes to any other aspects of the device. This is considered a substantial change, as the change may affect the quality and safety of the finished product.
- The manufacturer decides to change the sterilisation method for a medical device from gamma irradiation to ethylene oxide (EO) without changing any other aspects of the device. Assessment of the manufacturer’s sterilisation validation is required to ensure the minimum sterility assurance level (SAL) is achieved, and that EO residuals remaining on the medical device are below the threshold for acceptable levels. This is considered a substantial change.
- The manufacturer of a drug-eluting stent changes the supplier that provides the medicinal substance incorporated in the medical device. This is considered a substantial change. The manufacturer is required to provide evidence (such as Letter of Access for an existing Drug Master File and/or Certificate of Suitability) to support the quality and safety of the medicinal substance incorporated in the device, as well as documentation to support the manufacturer’s control over the new supplier.
- The manufacturer intends to change the sodium hyaluronate raw material specification to accommodate changes proposed by the raw material supplier. Validation test results have shown there is an impact on the quality, performance, and specification of the finished medical device. This is considered a substantial change
- A manufacturer intends to add a production line for a new category of Class 3 IVD medical devices that use nucleic acid technology (NAT) to detect infectious disease markers, whereas previously they have only been certified to produce immunoassay kits for detecting the same infectious markers. Even though the new NAT assays are the same ‘kind of device’ as the immunoassay kits that were previously assessed by the TGA, this is a substantial change to the manufacturing process.
- The manufacturer decides to transfer the manufacturing of drug-coated stents from an existing facility to a new manufacturing facility as an alternate measure to prevent loss of critical processes. This is considered a substantial change. Assessment of the manufacturer’s equipment and process qualification as well as transfer validation evidence is required to determine that the process transfer results in no impact to the quality, safety and performance of the finished medical device.
Product design – Medical devices (excluding IVD medical devices)
- The IFU for a pacemaker lead states that the medical device is safe for conditional use within MR-environments of up to 1.5T. The manufacturer performed additional testing on the device and test results have shown that the device can be conditionally used within MR-environments of up to 3T. The manufacturer would like to change the IFU to reflect this new information. Given that exposure of an implanted medical device to higher magnetic field strengths is likely to introduce new risks for the patient, this is considered a substantial change.
- The allowable upper storage temperature for a medical device has been increased due to new stability data becoming available. No other changes have been made to the device. This may affect device performance, and is therefore, considered a substantial change.
- Due to real-time aging data becoming available (or further accelerated-aging test results), the manufacturer of a drug-coated stent intends to extend the claimed product shelf-life from 12 months to 24 months. This is a substantial change to the packaging and shelf life of the medical device.
- The manufacturer of a coronary stent includes a new stent that has a larger diameter than the existing family of coronary stents. The stent diameter is outside the range of the previously approved variant range for the stents. As the larger diameter could significantly affect device related safety and performance, this is considered a substantial change.
- The manufacturer of a coronary stent increases the thickness of the wire in the stent to reduce the potential for stent fracture. The thickness is important to the performance of the stent and could affect the safety or efficacy of the device. Therefore, this is considered a substantial change.
- The manufacturer of a hemi-arthroplasty femoral stem/head changes the taper/bore angle of the device. This change is likely to affect the biomechanics of the joint replacement device and hence the performance of the device. Therefore, this is considered a substantial change.
- The manufacturer of a biological surgical repair mesh intends to add an additional manufacturing stage to provide surgeons with pre-cut meshes. This cutting work is currently being performed by surgeons in theatre during operations. This design change involves adding a manufacturing process that has not been previously assessed. Therefore, this is considered a substantial change.
- The manufacturer of a medical device intends to implement design changes that are driven by recent changes in the standards that were originally used to demonstrate compliance, or due to recent post-market data becoming available. Such changes are likely to affect the device quality, safety and performance. Therefore, this is considered a substantial change.
- During research and development processes, the manufacturer of an approved active implantable medical device has found new software algorithms that improve the devices’ function and usability. The new software algorithms are not limited to affecting the graphical user-interface of the device, only). This change requires new design validation and verification, and is therefore, considered a substantial change.
- Using previously approved manufacturing processes, the manufacturer of an intraocular lens (IOL) has made changes in the haptic geometry of the IOL and a change to the endotoxin specification limits (i.e. <0.2 Endotoxin Units per device) as recommended in the Medical Device Standards Order (Endotoxin Requirements for Medical Devices) 2018. However, the optical performance specifications of the device have not changed. The changes related to haptic geometry are likely to affect the device performance and interaction with internal tissues whilst changes to endotoxin limits impact the final product specification. This is considered a substantial change.
- The manufacturer of a balloon catheter changes the material of the polymer tubing used to manufacture the catheter from polymer A to polymer B and the manufacturer has not previously used polymer B (even if there were other similar devices on the market that use polymer B). The manufacturer would have no knowledge of the formulation or manufacturing processes of polymer B tubing in the competitors’ devices and would be required to assess the biocompatibility and performance of polymer B tubing in its own catheter. This is considered a substantial change. However, if the manufacturer has used polymer B tubing, with the same formulation and processing, in another previously approved catheter with the same type and duration of body contact and the same performance specifications then it may be acceptable to document the change to the QMS and update the technical file only. The updated technical files may be assessed/verified by the TGA during a subsequent on-site audit.
- The manufacturer of a cardiac catheter system decides to change the materials of the guide wire that is supplied with the system. As the guide wire would be in direct contact with the circulatory system, the change in material is likely to affect the biocompatibility of the medical device. In addition, the quality and safety of the new materials require assessment and any impact on performance should be considered. This is considered a substantial change.
Product design – IVD medical devices
- A manufacturer changes the design of an IVD medical device for detecting human immunodeficiency virus types 1 & 2 (HIV 1 & 2) to meet less stringent performance specifications. The change affects both sensitivity and specificity. Although the change does not alter how the device is used, or require labelling changes, it does alter the performance characteristics of the assay when compared to those established under previous clinical performance studies. This is considered a substantial change.
- A change in the IFU for a Class 4 IVD medical device, to include a new anticoagulant type for plasma samples that are to be tested using the device, or any other (new) sample type not previously assessed, is considered a substantial change. This also applies to an extension of the intended purpose, to include testing of cadaveric samples (i.e. samples that have been collected post-mortem from a potential organ donor) to detect the presence of infectious agents).
- An IVD manufacturer holding a TGA-issued design examination certificate for Class 4 Immunohaematology reagents (IHRs) makes changes to the existing kind of device to include additional reagents not previously listed on the certificate. This substantial change requires submission of an application to assess the new IHRs, and to add them to the certificate.
- An IVD medical device manufacturer makes a material change to a reagent (for example, changing a monoclonal antibody used in an ELISA kit, or the RNA primers included in a NAT assay) and the risk assessment shows that this could result in a change to the performance of the device outside the performance specifications already approved by the TGA. This change has the potential to affect clinical decision-making, and is therefore, considered a substantial change. Note: If the manufacturer conducts validation studies to demonstrate that the change does not impact the performance of the IVD medical device, the manufacturer will still need to submit a substantial change notification and application so that the TGA can review manufacturer’s validation. This applies regardless of whether the studies demonstrate there has been no impact on performance of the IVD medical device.
- A manufacturer has previously conducted stability validation studies for a new Class 4 IVD medical device by periodically testing kits selected from three production lots that have been stored under recommended storage conditions for a period of 13 months. The manufacturer has generated sufficient data to demonstrate that performance remains consistent throughout the specified storage period and approval was granted to the manufacturer to assign a 12-month shelf life to the kits.
The manufacturer continued to monitor the ongoing stability studies for a further period of 6 months as outlined in the existing stability protocol, and sufficient real time stability data generated over the cumulative storage period of 19 months under the recommended storage conditions is available. The results continue to meet the assigned acceptance criteria throughout the storage period; therefore the manufacturer may assign a longer shelf life of 18 months based on the extension of testing under the existing stability protocol. As part of the ongoing stability-monitoring program, the manufacturer should also periodically monitor the performance of the product at the end of the assigned shelf life to ensure routine production batches continue to meet the required specifications.
The extension of shelf life is considered a substantial change. However, if there were no changes to the protocol or the assigned acceptance criteria, the TGA may determine that no further assessment is required. In this case, no assessment fees will be charged, and a new version of the certificate with the changes reflected on the certificate history will be issued.
Information to be provided with medical devices, including IVD medical devices
- The IFU for a medicated balloon catheter is updated to provide instructions on how to access different vessel types that were not previously addressed in the labelling or IFU. This would change the intended use of the medical device and the techniques used during the procedure, and is therefore, considered a substantial change.
- The manufacturer removes a contraindication for a medical device in the IFU, without making any changes to the device. This change effectively broadens the indications for the device, and is therefore, considered a substantial change. This is also the case if a manufacturer adds a new (previously unapproved) indication to the IFU.
- The manufacturer removes a precaution and/or warning previously stated for the medical device, or IVD medical device in the IFU, without making any changes to the device. The purpose for removal of the precaution and/or warning requires careful consideration to determine the impact on the overall risk-benefit profile of the device. This change requires submission of substantial change notification and application so that the TGA can review Therapeutic Goods Administration Changes affecting TGA-issued conformity assessment certificates V2.0 August 2021 Page 28 of 33 the manufacturer’s amended risk documentation and updated IFU. The TGA’s assessment of this change may not require extensive review.
- A change in labelling and/or IFU of the medical device to add a statement that the device is non-pyrogenic or endotoxin free, and is therefore, considered a substantial change.
- The manufacturer decides to change the Unique Product Identifier (UPI) of a medical device, or IVD medical device, without making any other changes, except to the device labelling and IFU to reflect the new name. This change requires submission of substantial change notification and application so that the TGA can verify the manufacturer’s updated labelling and IFU, and if required update the name of the device listed on the design or type examination certificate. The TGA may determine that no further assessment is required. In this case, no assessment fees will be charged, and a new version of the certificate with the changes reflected on the certificate history will be issued.
- After clinical investigations, the manufacturer of a dermal filler decides to add recommendations for use of a certain type of cannula in their IFU as part of the surgical procedure. The manufacturer clarifies that this cannula will not be packaged together with the product. However, this change is likely to be substantial since the new operation procedure or technique will involve specific training and verification.
- The manufacturer decides to change the form of the IFU supplied with the device intended for professional use, from paper format to electronic or online format. For medical devices that are supplied sterile, removal of the paper form of an IFU from inside a sterile packaging unit could result in changes to the bioburden and sterilisation dosing requirements established for the device. If it is determined as part of the manufacturer’s risk assessment process, that when changing to supply of an electronic IFU the effectiveness of any mitigation steps applied cannot be fully verified, this is considered a substantial change.
Appendix B: Examples of changes that do not require an application for substantial change
Note that the examples are not all-inclusive and may not be applicable in all cases4.
As outlined in Sections 4–5, change of this nature described in this section will require the manufacturer to maintain documentation of the changes including records of the details and analysis of changes and should be made available to the TGA for review, upon request or at the next surveillance audit.
Changes to medical devices, including IVD medical devices, that relate to QMS, production and product design
- The manufacturer changes the supplier of the polyethylene packaging for their sterile medical devices; the packaging configuration and specifications remain the same. The manufacturer uses the same packaging integrity test protocols as that previously approved and an analysis shows the new packaging has no impurities that could affect the devices’ biocompatibility. This is not likely to require notification to the TGA. However, it is still a requirement of the Regulations that after any change the manufacturer must ensure its devices continue to meet the requirements of their QMS and the essential principles. Documentation of the change would be required and may be followed up for review at a subsequent TGA QMS audit. The Technical file for the product should also be updated.
- The manufacturer decides to add certified foreign language translations of the labelling and IFU for use in other regulatory jurisdictions, while no change is to be made to the content of the approved product labelling. Since this change is unlikely to introduce new risks associated with the use of the medical device, or IVD medical device, notification to the TGA would not be necessary, but the change should be recorded in the QMS documentation.
- The manufacturer of a range of coronary stents intends to begin manufacturing a new stent with a different diameter to those already approved for that stent range. The new stent diameter falls within the previously approved variant range and there are no other changes (i.e. to length of stent). The manufacturer has determined that the additional stent diameter does not represent the worst-case for any physical parameters that require testing, and the risk assessment for the change indicates there are no new risks being introduced with the additional size variant and therefore it has no impact on the safety or performance of the device. This is not considered a substantial change; however, the product technical file is required to be updated, and made available upon request. Note, the manufacturer may be required to submit a Device Change Request application to the TGA, to update the Australian Register of Therapeutic Goods (ARTG) certificate so that it accurately reflects the number of variants supplied in Australia for that kind of medical device.
- The manufacturer of a hip joint implant intends to reduce the manufacturing tolerances allowable during production, enabling them to manufacture the medical devices closer to their design specifications without introducing new design features. Since the tightening of acceptance criteria is not expected to introduce any new risks, the change is not likely to be considered a substantial change. However, it is still a requirement of the Regulations that for any change, the manufacturer must ensure the device continues to meet the essential principles. Documentation of the change would be required and may be followed up for review at a subsequent TGA QMS Inspection. The Technical file for the product should also be updated.
- A manufacturer extends the shelf life of, for example, a Class IIa, non-sterile medical device, or a Class 2 IVD medical device that is covered by a TGA Schedule 3, Part 1 certificate. The extension of shelf-life is carried out under the manufacturer’s QMS change control procedures. This change does not require notification to the TGA however, it is still a requirement of the Regulations that for any change, the manufacturer must ensure the device continues to meet the essential principles. Documentation of the change would be required and may be followed up for review at a subsequent TGA QMS Inspection. The Technical file for the product should also be updated.
- A manufacturer intends to make a change to a generic reagent (e.g. buffer) included in the formulation of an IVD medical device as a result of a change in supplier. The raw material specifications for the replacement reagent are identical to the previous reagent. Results of the risk analysis and the validation and verification studies conducted by the manufacturer indicate that there is no impact on performance and therefore no new safety or performance concerns. The manufacturer should document the changes and relevant tests in the QMS and the technical file for the product should be updated.
Information provided with a medical device, including IVD medical devices
- The manufacturer of a low risk IVD medical device has updated the information supplied with a device to include a new limitation relating to the performance of the device when used on patients who have been treated with a new medicine, which could interfere with the result. The medicine does not affect the indications for use for the device, nor significantly affect the risk profile. The manufacturer should document the change in the QMS and the technical file for the product should be updated.
- The manufacturer rebrands or renames a Class 2 or 3 IVD medical device covered under a device category listed on the manufacturer’s TGA Schedule 3, Part 1 conformity assessment certificate. The change is required to be documented under the manufacturer’s QMS. However, an application for a change to an existing certificate is not required to be submitted unless there are additional changes to the device that impact the intended use or intended user of the device.
Appendix C: Examples of certificate transfers
Examples below illustrate some events that would be considered to trigger the transfer of a TGA-issued conformity assessment certificate.
- A manufacturer holds a TGA-issued conformity assessment certificate for the manufacture of several Class 4 IVD medical devices. The company also manufactures a range of lower class IVD medical devices that are not included within the scope of their QMS certificate. The manufacturer decides to separate the Class 4 IVD medical devices from the rest of the business through sale of the lower class IVD medical devices to another company. The Class 4 IVD medical device manufacturer retains the same QMS but changes its name. In such a case, Regulation 4.8 would require the manufacturer to notify the TGA about the change of name within 3 months.
- A corporation has multiple divisions, some medical device (or IVD medical device) and some pharmaceutical. Each division maintains its own QMS. The corporation decides to separate the pharmaceutical and device businesses into two separate entities. The pharmaceutical business is to retain the current business name and registration. The device businesses are to change name and register as new business entities. The device businesses hold TGA conformity assessment certificates for the manufacture of the devices. The certificates are transferred to the new entity. In such a case, Regulation 4.7 would require the new business to apply to the TGA for the name of the manufacturer to be changed on the conformity assessment certificate within 3 months.
- A manufacturer holds a TGA-issued Conformity Assessment Certificate and amalgamates with another corporation that manufactures medical devices (or IVD medical devices) that do not require a TGA-issued Conformity Certificate for supply in Australia. The new entity (business) takes on a new name but retains all the current manufacturing sites of the two prior corporations. The TGA-issued conformity assessment certificate is transferred to the new entity, but the scope of the certificate does not increase to cover the devices at the second site. In such a case, Regulation 4.7 would require the new business to apply to the TGA for the name of the manufacturer to be changed on the conformity assessment certificate within 3 months.
- A manufacturer holds a TGA-issued conformity assessment certificate and declares bankruptcy. The Trustee in bankruptcy becomes the legal manufacturer of the medical devices (or IVD medical devices). In such a case, Regulation 4.6 would require the trustee to inform the TGA of the event within 3 months.
Footnotes
- See subsection 41EJ(3) of the Act.
- Under section 41EB of the Act, an application is not effective if the prescribed application fee has not been paid. The fee for an application for conformity assessment certificate is prescribed in Schedule 5, Item 1.1 of the Therapeutic Goods (Medical Devices) Regulations 2002.
- Some of the examples below have been modified from the U.S. Food and Drug Administration guidance: Deciding When to Submit a 510(k) for a Change to an Existing Device: Guidance for Industry and Food and Drug Administration Staff (October 2017).
- Some of the examples below have been modified from U.S. Food and Drug Administration guidance: Deciding When to Submit a 510(k) for a Change to an Existing Device: Guidance for Industry and Food and Drug Administration Staff (October 2017).
Page history
Title changed from 'Changes affecting TGA-issued conformity assessment certificates' to 'Changing or transferring conformity assessment certificates ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Title changed from 'Changes affecting TGA-issued conformity assessment certificates' to 'Changing or transferring conformity assessment certificates ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.