Meeting the eligibility criteria for orphan drug designation
Guidance on how to prepare your application for orphan drug designation so that it meets eligibility criteria and requirements.
Purpose
This guidance is for sponsors preparing an application for orphan drug designation. It covers eligibility criteria and supporting documentation.
For information about the whole process of orphan designation, refer to Applying for orphan drug designation of a prescription medicine.
Read the guidance on the Prescription medicines registration process for assistance with registration.
Legislation
Eligibility for orphan drug designation
Consider all of the eligibility criteria below before you lodge a designation application.
Orphan drug designation may be granted for:
- a previously unregistered medicine
- an already registered medicine with a new orphan indication, a new dosage form medicine or a major variation application that meets all relevant criteria including the significant benefit criterion.
Eligibility criteria for the Orphan Drug designation
A medicine, including vaccines or in vivo diagnostic agents, may be eligible for orphan drug designation if all orphan criteria in the table below are satisfied (regulation 16J of the Therapeutic Goods Regulations 1990- external site [the Regulations]).
In addition to the standard orphan designation, a pathway to seek orphan designation for new dosage form medicines is available. Eligibility for new dosage form medicines is intended to provide an incentive to sponsors to register medicines on the Australian Register of Therapeutic Goods (ARTG) that introduce a new dosage form that would not be financially viable in the absence of a TGA fee waiver. An example of a new dosage form medicine is a new oral liquid dose form (e.g. a syrup), where a tablet is already registered.
Combinations of medicines that are not fixed dose combinations require separate applications (one for each component of the non-fixed dose combination).
Orphan drug designation
Orphan drug designation for a medicine is specific to the:
- sponsor
- orphan indication for which designation was granted
- dosage form of the medicine.
Orphan designation cannot be transferred from one sponsor to another.
Orphan designation for multiple indications
The application can be made for only one indication of the medicine. If you seek orphan drug designation for more than one indication, separate designation applications need to be submitted for each orphan indication. In this regard, 'treatment' and 'prevention' or 'diagnosis' of the same condition are considered as separate indications and should be the subject of separate applications for designation.
Supporting documentation requirements
The main body of your designation application should be no more than 30 pages. Additional supporting documentation can be included as attachments.
The main body should address:
- steps 1 to 5 of this guidance document including a description of the condition and justifications for all relevant eligibility criteria
- where applicable, a justification as to why a new dosage form may be necessary for the Australian population.
Attachments should include:
- summaries of pivotal studies, e.g. the study synopsis included as part of the body of the clinical study report, or where study synopses are not available, a summary of the study with sufficient detail to inform assessment (a full module 2 summary is not required); do not submit full study reports
- supporting evidence based on clinical trial data. Comparator studies are expected to be generated (pivotal study reports)
- summaries of any available other important safety data obtained in the preclinical and clinical setting (both in the orphan condition and in other conditions and/or how safety information from other formulations may be applicable)
- where published papers are highly relevant, the full text of such literature (including supplementary appendices)
- other forms of literature references or unpublished reports and expert statements may also be used in addition to the pivotal study summaries but would be considered low level evidence
- an abbreviations list
- it is expected that any supporting information is currently relevant. Information that is out of date or obsolete must not be submitted
- applications for designation which are based on the financial viability of the medicine are considered under financial viability.
EMA Orphan Drug designation applications supported by an Australian specific annex will be considered if the content is current and relevant.
The Australian specific annex should include:
- updated prevalence or justification why EMA prevalence applies
- financial viability for Australia
- comparison against ARTG registered therapeutic goods/justification of significant benefit
- details of the regulatory status in Australia and overseas
- pivotal study summaries.
Content of the designation application
We will determine the validity of the justifications on a case by case basis. As part of the routine designation process, the extent to which criteria are met will be assessed, at the time a decision is made on the application
A designation application may be withdrawn by the sponsor prior to decision. The designation may be revoked following decision if the TGA is satisfied that any criterion for orphan designation is no longer met (Regulation 16M). We will not routinely review designation decisions during the period of their validity but may do so in exceptional circumstances.
Step 1: Provide a description of the condition
Details of the orphan condition
The condition proposed by the sponsor is the starting point for the scientific evaluation.
Your designation application must provide the details of the condition that the medicine is intended to diagnose, prevent or treat. This information should include:
- a clear description of the disease or condition in question
- details of the causes and symptoms
- refer to the condition according to international disease classification system such as the WHO's International Classification of Disease (ICD) or other well recognised systems of classification (e.g. WHO central nervous system tumour classification), where applicable.
The TGA may consult the sponsor if we do not agree with the original proposed orphan condition, taking into account the available data to determine the proposed orphan condition (e.g. we may consider that it is broader than the sponsor's proposal).
Proposed orphan indication
The application must be for one indication only. You must submit details of the proposed orphan indication:
- define the target condition or disease distinguishing between treatment, prevention and diagnostic indications, including the patient group proposed where applicable
- the orphan indication may comprise a broader population than the population defined by the proposed therapeutic indication
- the orphan indication is the population on which the prevalence is estimated
- include the ATC code where it has been assigned.
Please note that the TGA may require the sponsor to modify the indication during the designation process.
Proposed orphan indication and therapeutic indication at registration
The medical condition that the designated orphan indication treats, prevents or diagnoses is published on the designation notices page and is not always the same as the therapeutic indication that is approved and registered on the Australian Register of Therapeutic Goods.
The therapeutic indication is the proposed indication for ARTG registration, based on the proposed indication at the time of the designation. The medical condition specified in the orphan designation is often broader than the therapeutic use ('indication') for which the medicine is eventually registered.
This is because the therapeutic indication at registration is based on the evidence that the company provides in the registration application. Generally, the registered therapeutic indication is based on the characteristics of patients in the clinical trials and might exclude certain groups like the very young or pregnant women, or it might include patients with only one variant of the condition.
For example, a medicine originally designated orphan for cystic fibrosis might subsequently be registered for use only in those aged over 12 years with a specific mutation (change) in the CFTR gene (reflecting studies showing positive outcome in this group), rather than for all patients with cystic fibrosis.
Medical plausibility
Clinical data are required to support the rationale for the development of the orphan medicine in the proposed condition.
There are two aspects to 'medical plausibility':
- the rationale for use of the medicine in the proposed orphan indication (the medicine will be effective in the treatment, prevention or diagnosis of the condition)
- if the proposed orphan condition is a subset of a condition affecting a larger population, the medicine would not be effective for the larger population.
The section should include:
- active substance: description of the medicine
- the pharmacological class
- the mechanism of action
- details of the rationale for the use of the medicine in the orphan indication
- where the proposed orphan indication refers to a subset of a particular condition, a justification for restricting the use of the product to the subset
- typically, only recognised distinct medical entities would be considered as valid conditions for designation, while sub-sets would only be exceptionally considered.
When considering the definition of 'condition ' and what constitutes a 'valid condition' as opposed to what would be considered 'invalid subsets of a condition', take into account the following:
- recognised distinct medical entities that are defined in terms of their specific characteristics (e.g. pathophysiological, histopathological, clinical characteristics) would generally be considered valid conditions
- the characteristics defining a distinct condition should determine a group of patients in whom development of a medicine is plausible, based on the pathogenesis of the condition and pharmacodynamic evidence and assumptions, the dosage form if this is specific for a group of patients
- patients may be affected by more than one condition. Generally, the intersection of two (or more) concomitant conditions would not be considered as a valid condition. However, it could be acceptable, if such intersection resulted in a certain new evaluable characteristic essential for the pharmacological effect and the medical outcome
- where the proposed orphan indication refers to a subset of a particular condition, a justification for restricting the use of the product is required. The methods or criteria used to delineate this population subset must be described
- a subset of a disease which, when considered as a whole has a prevalence greater than 5 in 10,000, could be considered a valid condition if patients in the subset should present distinct and unique evaluable characteristics with a plausible link to the condition and such characteristics should be essential for the product to carry out its action
- subgrouping based on the age of the sub-population would only be considered appropriate where the product would be ineffective in the remaining population
- paediatric indications will be considered eligible subsets for orphan designation where the prevalence criterion is met in relation to the whole of the disease (i.e. including adults), or is met where the disease is different in adults, or specific to the paediatric subgroup
- different degrees of severity, stages of a disease or lines of therapy (e.g. second line treatment) would generally not be considered as distinct conditions.
Use of biomarkers to subset a condition
- Personalised medicines require the use of biomarkers to identify the specific variant of the disease. However, the use of biomarkers to identify a subset of patients for whom the medicine can be used is generally not accepted for orphan designation and would only be considered as an exception.
- The subset proposed should fall entirely within a distinct medical condition (therefore not spanning populations affected by more than one distinct medical entity)
- Sub-setting a condition with the use of biomarkers will not be acceptable unless the sponsor provides solid scientific evidence that the activity of the product would not be shown on the larger population. The genetic subtype/profile and pathophysiological characteristics associated with the subset should be so closely linked to the diagnostic and/or preventive and/or treatment action of the product that the absence of these characteristics will render the product ineffective in the rest of the population suffering from the condition. In exceptional circumstances evidence of no ongoing phase I/II or III studies would be acceptable to demonstrate lack of activity.
- Further information on the use of biomarkers in the context of orphan designation is available in a 2014 publication on the Use of biomarkers in the context of orphan medicine designation in the EU (PDF, 204 Kb)- external site.
Step 2: Provide a justification of the life-threatening or seriously debilitating nature of the condition
The severity of the disease, i.e. its seriously debilitating or life-threatening nature needs to be justified, based on objective and quantifiable medical or epidemiological information.
Justify the:
- life-threatening nature of the condition based on figures of mortality and life expectancy
- seriously debilitating nature of the condition based on morbidity over the course of the disease and its consequences on patients' day-to-day functioning.
Serious debilitation, or fatal outcome should be a prominent feature of the target disease and therapeutic indication, i.e. affect an important portion of the target population.
Step 3: Provide either the prevalence of the condition or a justification for lack of financial viability
Prevalence of the proposed orphan condition
Where orphan drug designation is sought on the basis of the prevalence of the orphan condition or disease in Australia the following information should be provided:
- orphan condition prevalence calculated for the condition as applied for in the designation application
- the methodology for the calculation should be clearly described.
Prevalence calculation for medicines for treatment
Where medicines are intended for treatment, you should provide information on the prevalence in the number of persons with the disease or condition which the medicine may treat at a specified instant in time, in a given population affected by the condition in Australia, at the time of designation application.
Prevalence calculations for medicines for diagnosis or prevention
For medicines intended for diagnosis or prevention of a condition, the prevalence calculation should be based on the population to which the product could be administered on an annual basis in Australia. For example, following successful vaccination campaigns, although the vaccinated population is very large, the prevalence of the condition in question may be very low. The prevalence calculation in these cases should be based on the population vaccinated on an annual basis. Likewise, a diagnostic test for a rare condition should be based upon the annual usage of the test.
Prevalence and incidence of the condition in Australia
Where designation based on 'lack of financial viability' is sought, information on the prevalence and incidence in Australia of the condition at the time at which the application for designation is made should be provided. The medicine should not generally be intended for use in other conditions. If there are other conditions for which this medicine is intended these should be stated for consideration (see financial viability for further information).
Required prevalence reference documentation
You should consider the following when seeking orphan designation on the basis of prevalence:
- The documentation should include appended authoritative references which demonstrate that the disease or conditions for which the medicine would be administered, affects less than five in 10,000 persons in Australia at the time at which the application for designation is submitted, where these are available.
- The documentation should include or refer to a review of the relevant scientific literature and shall provide information from relevant databases in Australia (e.g. the Australian Bleeding Disorders Registry), where these are available. In the absence of epidemiological data or databases and when only case reports of the disease are available in Australia, reference may be made to epidemiological data and databases available overseas, supplemented with an explanation of the extrapolation to the Australian population.
- You should clearly explain how the estimated proposed orphan condition prevalence has been calculated, indicating the methods and results for identifying source data/documentation (published references and databases) and calculating the prevalence. Studies should be summarised in tabular format including all relevant information such as definition and size of the study population and case definition etc.
- Where a disease or condition has been considered within the framework of other programs or activities on rare diseases, this information should be provided. In the case of diseases or conditions included in projects financially supported in Australia or internationally in order to improve information on rare diseases, a relevant extract from this information, including in particular, details of the prevalence of the disease or condition in question, should be provided.
- If up-to-date references (published references and databases) are not available, you should provide a clear basis for the assumption that the disease or condition will meet the orphan condition prevalence criteria.
Financial viability
In the case of applications for designation which are based on the financial viability of the medicine a justification for the lack of financial viability must be provided.
To support an application for Orphan Drug designation on the basis of the 'not financially viable' criterion, it is the responsibility of the sponsor making the application to provide the TGA with sufficient evidence to allow a delegate to be satisfied that the criterion has been met.
We will look at financial viability of a medicine and not for a particular indication or formulation of that medicine. New dosage forms will only be considered if the medicine is in a new dosage form that is currently not registered on the ARTG, for a serious or life-threatening condition with established significant benefit. New dosage forms will not be considered where an application is made on the basis of prevalence only.
Your designation application should include information about costs incurred or expected to be incurred in the course of developing, producing, marketing and supplying the product. These costs should include, but are not limited to, clinical studies, formulation studies, stability studies, and the costs of producing, marketing and supplying the product in Australia.
In addressing the above, the marginal or incremental costs of manufacturing and marketing the product in Australia should be used not the full international cost, and supporting evidence provided.
A guide to the information and supporting documents that should be provided follows.
Grants and tax incentives
The documentation provided should include details of any grants, tax incentives or other incentives received in Australia that will offset Australian costs.
Past and future costs
You should include a statement of and justification for all development and marketing costs (in Australian Dollars) for the product that the sponsor has incurred or expects to incur to bring the product to the Australian market. If the local Australian sponsor is expected to contribute to total global development costs this should be itemised separately, and you should also provide a clear method and justification of the method of determining contribution to total global development costs.
Regardless of whether the product is already authorised for any indication or not, for the purpose of the 'not financially viable' criterion, the total marginal cost of bringing the new product (and not just a particular indication or formulation alone) should be provided, by year.
Production and marketing costs
You should provide a statement of and justification for all production, marketing and other costs for the intended Australian product that the sponsor has incurred or expects to incur.
Expected revenues
You should provide an estimate of and justification for all the Australian revenue for the product that the sponsor expects to earn during the first 10 years following authorisation. This section should include explanations of any impediments to achieving commercially viable sales or revenue. You should assess the potential for PBS reimbursement and appropriate assumptions should be made in forecasting revenue.
Certification
You should ensure that all cost and revenue data are determined in accordance with generally accepted accounting practices and the information in support of the claim must be submitted under the signature of the Chief Financial Officer or equivalent of the sponsor who is a qualified accountant in Australia.
Step 4: Comparison against registered therapeutic goods for diagnosis, prevention or treatment in Australia
You must establish that there are no therapeutic goods included on the Australian Register for Therapeutic Goods for diagnosis, prevention or treatment of the condition in question, or if such therapeutic goods exist that the medicine will bring improved efficacy, safety or a major contribution to patient care to those affected.
Any reference to a registered therapeutic good must be limited to the relevant register entry. Therefore, a product that is administered or applied outside the registered approved use ('off-label' use) cannot be considered registered therapeutic good for the purposes of the Regulations.
Note that where a medicine is already registered for the orphan indication, the medicine would need to demonstrate a significant benefit over itself (e.g. through a new dosage form).
Registered therapeutic goods
You must review registered therapeutic goods for diagnosis, prevention or treatment of the proposed indication in Australia and provide:
- details of any registered therapeutic goods for diagnosis, prevention or treatment for this indication (Overview table of Tradename(s), holder of the ARTG entry, and the registered indication)
- either a justification demonstrating significant benefit over any registered therapeutic goods, or a declaration that there are no registered therapeutic goods in Australia in accordance with ARTG entries on the date of designation lodgement.
Justification of significant benefit
When the orphan drug designation application is based on significant benefit, a comparison with registered therapeutic goods is required for designation, as opposed to applications for conditions where there are no therapeutic goods are registered for diagnosis, prevention, or treatment.
Justification of significant benefit should be based on the available evidence at the time of the designation lodgement. Suitable evidence must be provided demonstrating the benefit of the medicine to the population with the condition. Significant benefit may not be based on a combination of registered medicines (where this is not a fixed dose combination).
You must establish significant benefit as compared with all therapeutic goods that are included on the ARTG at the time of designation application lodgement (including the medicine itself). If new goods for the diagnosis, prevention or treatment of your proposed indication are included on the ARTG after you lodge your application, you will have the opportunity to submit a further justification of significant benefit in relation to those goods before a decision is made on your application.
We will not assess significant benefit against comparators that are the subject of a concurrent designation application. For a claim of 'significant benefit', we will evaluate whether there is a high probability for patients to experience a clinically relevant benefit.
Any claim has to be supported by solid evidence. Supporting evidence should be based on clinical trial data:
- Comparator studies are expected to be generated (pivotal study reports).
- Scientific argument/justification for the significant benefit of the medicine relative to products not studied in available clinical trials (this may involve cross study comparisons).
- Justifications or comparator studies are required for all identified registered products.
The evidence must be considered in the light of the particular characteristics of the condition (life expectancy, symptoms and quality of life) and all registered therapeutic goods for diagnosis, prevention, or treatment. Evidence will be weighted based on the strength and clinical relevance of the supporting data. The justification could include benefits and risks based on traditional safety and efficacy endpoints and other health outcomes.
The justification of significant benefit should establish one of the following:
- Improved efficacy - for the entire applicable population suffering from the proposed orphan condition
- Improved safety - a better safety profile or a better tolerability for the entire applicable population suffering from the proposed orphan condition
- Major contribution to patient care:
- ease of use in a particular patient group or self-administration, e.g. if the new treatment allows ambulatory treatment instead of treatment in a hospital only or if it has a significant impact on convenience of use and reduces treatment burden, or
- significantly improved adherence to treatment due to a change in pharmaceutical form (e.g. modified release formulation), provided there are documented difficulties with the existing dosage form and data showing better clinical or patient outcomes with the new dosage form. Difficulties should be documented in peer-reviewed publications, patients' registries, therapeutic guidelines or dispensing of compounded medication or other applicable data. A better clinical outcome could include better quality of life.
'Significant benefit' should not be based on:
- possible increased supply/availability due to shortages of registered medicines
- enhancement of the pharmaceutical quality of a product or
- a new pharmaceutical form, a new strength or a new route of administration, unless it brings a major contribution to patient care or
- an alternative mechanism of action in the absence of a favourable comparison to registered therapeutic goods.
Step 5: Description of the stage of development
Summary of the development of the product
You should briefly describe the current development status of the proposed orphan medicine, clinical investigation and details of the proposed registration application dossier. You should supply information on any proposed developments and registrations in other indications or other patient groups. The full study reports of non-clinical and clinical studies undertaken need not be provided for the designation.
Details of current regulatory status in Australia and overseas
You must include a summary of the world-wide regulatory status and marketing history (including dates) for the medicine, which details (where applicable):
- current investigational uses
- previous marketing approvals, including details of the indications for which the medicine is approved overseas
- orphan status
- a copy of the EMA decision letter where applicable
- adverse regulatory actions (if any) in any country.
This section should include details of whether orphan status has been applied for or granted in other countries with respect to the medicinal product. If orphan status has been granted elsewhere, it is useful to attach a copy of the decision on Orphan Drug designation to the application.
Refusal to approve on grounds of safety
You must outline whether or not any of the regulatory agencies listed below has refused to approve the medicine for reasons related to the medicine's safety. A medicine is not eligible for Orphan drug designation if any of the following regulatory agencies have refused to approve it for use for the condition for a reason related to the medicine's safety:
- TGA
- United States Food and Drug Administration (US FDA)
- the European Medicines Agency (EMA)
- Health Canada
- the Medicines and Healthcare Products Regulatory Agency of the United Kingdom.
Definitions
The following definitions are applicable in the context of this guideline:
- Condition: any deviation(s) from the normal structure or function of the body, as manifested by a characteristic set of signs and symptoms (typically a recognised distinct disease or a syndrome)
- Orphan Condition: a condition that meets the Orphan Drug criteria specified in the Therapeutic Goods Regulations 1990.
- Orphan indication: the proposed indication for the purpose of Orphan Drug designation. This specifies if the medicine which is the subject of the designation application is intended for the diagnosis, prevention or treatment of the Orphan Condition.
- Therapeutic indication: the proposed indication for the ARTG registration, based on the sponsor's proposal.
- Medicine:
- therapeutic goods (other than biologicals) that are represented to achieve, or are likely to achieve, their principal intended action by pharmacological, chemical, immunological or metabolic means in or on the body of a human; and
- any other therapeutic goods declared by the Secretary, for the purpose of the definition of therapeutic device, not including therapeutic devices.
- Life threatening: a prominent feature of the condition (i.e. affecting an important portion of the target population) is serious illness from which death is reasonably likely to occur within a matter of months, or from which premature death is reasonably likely to occur in the absence of treatment based on mortality and life expectancy data.
- Seriously debilitating condition: a prominent feature of the condition (i.e. affecting an important portion of the target population) is morbidity with a well-established, major impact on the functioning of the person based on objective and quantifiable medical or epidemiological information. Short lived and /or self-limiting morbidity is not considered seriously debilitating
- New dosage form medicine: means a medicine that
- has the same chemical, biological or radiopharmaceutical active ingredient (or fixed combination of such ingredients) as another medicine that is included in the Register
and- has an indication in common with that other medicine; and
- does not have the same dosage form as that other medicine.
- has the same chemical, biological or radiopharmaceutical active ingredient (or fixed combination of such ingredients) as another medicine that is included in the Register
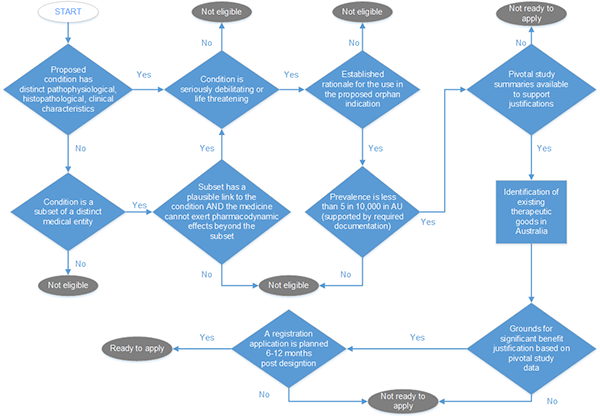
Flowchart guide to determine eligibility for standard orphan designation
Excluding financial viability and new dosage form medicines.
Flowchart guide to determine eligibility for standard orphan designation
{kind=link}
A flowchart depicting a decision-making process for determining eligibility for orphan drug designation. The flowchart consists of diamond-shaped decision points and oval endpoints, connected by arrows labelled "Yes" or "No".
The process flows as follows:
1. Start
2. "Proposed condition has distinct pathophysiological, histopathological, clinical characteristics?"
If No: "Condition is a subset of a distinct medical entity?"
If No: "Not eligible"
If Yes: Proceed to next step
3. "Condition is seriously debilitating or life threatening?"
If No: "Not eligible"
If Yes: "Subject has a chronically debilitating condition AND life expectancy cannot used pharmacodynamic effects beyond the acute?"
If No: "Not eligible"
If Yes: Proceed to next step
4. "Established scientific principles support orphan indication?"
If No: "Not eligible"
If Yes: "Patient population in Australia < or = to 5 in 10,000 (AU) (supported by required documentation)?"
If No: "Not eligible"
If Yes: Proceed to next step
5. "Pivotal study demonstrated a clinically relevant effect to support registration?"
If No: "Not ready to apply"
If Yes: "Identification of existing therapeutic options in Australia"
6. "Grounds for significant benefit justification based on clinical data?"
If No: "Not ready to apply"
If Yes: "A registration application is planned in 6-12 months post designation?"
If No: "Not ready to apply"
If Yes: "Ready to apply"
The flowchart illustrates a step-by-step evaluation process, with multiple criteria that must be met to reach the "Ready to apply" outcome.
References
Parts of this guidance document are derived from the following documents:
Footnotes
- The word 'condition' is used in the text of the Therapeutic Goods Regulations 1990. This is intended to ensure that the regulation applies also to treatments for conditions which are not classical diseases, in particular genetic disorders
Page history
Title changed from 'Orphan drug designation eligibility criteria' to 'Meeting the rules for orphan drug applications' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Update to currency of data required for criteria assessment.
Updates to align with commencement of the provisional approval pathway.
Updates to provide additional information on the use of biomarkers.
Minor updates and editorial changes.
Original publication.
Title changed from 'Orphan drug designation eligibility criteria' to 'Meeting the rules for orphan drug applications' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Update to currency of data required for criteria assessment.
Updates to align with commencement of the provisional approval pathway.
Updates to provide additional information on the use of biomarkers.
Minor updates and editorial changes.
Original publication.