Understanding personalised medical devices rules (including 3D-printed devices)
Guidance and examples to help you understand the regulatory framework for personalised medical devices.
Purpose
This document provides information and examples to help you understand the Australian regulatory framework for personalised medical devices (the Framework) and provides you with information and examples about how these devices are regulated, and what your regulatory obligations are if you are manufacturing or supplying a personalised medical device.
The intention of the examples is not to tell you how every device is regulated, but to provide enough information to help you identify the concepts and boundaries in the Framework so you can apply these concepts to your own circumstances.
A decision tree has been included at Appendix 1 to help manufacturers and sponsors of medical devices identify how their personalised medical devices will be regulated under the Framework.
Background and overview
Personalised medical devices are devices that are either designed and manufactured, or modified after they are supplied, to suit an individual.
Other resources
The TGA understands that many manufacturers and suppliers of personalised medical devices are not familiar with the general principles and elements of medical device regulation. There are many resources available to assist you with understanding how medical devices are regulated in Australia including:
- The Australian Regulatory Guidelines for Medical Devices: a range of information relevant to anyone who manufactures, imports, exports or otherwise supplies medical devices in Australia.
- The medical device inclusion process guidance: a step-by-step guide to applying to the TGA for regulatory approval of a medical device.
- SME Assist: a dedicated service that the TGA offers to help small-to-medium enterprises (SMEs), researchers, start-ups and those unfamiliar with regulation to understand their legal obligations. The SME Assist website contains a range of resources to assist people who have not had dealings with the TGA before, including an overview of medical device regulation.
- The Medical Devices Information Unit (MDIU) are available to assist you with specific enquiries Monday to Friday, 8:30am – 5:00pm (AEST). You can contact the MDIU on 1800 141 144 or via devices@tga.gov.au.
Patient-matched medical devices
The definition of a patient-matched medical device, as set out in the Regulations, is as follows:
patient-matched medical device means a medical device that:
is manufactured by the manufacturer, within a specified design envelope, to match:
either or both of the anatomical and physiological features of a particular individual; or
a pathological condition of a particular individual; and
is designed by the manufacturer (even if the design is developed in consultation with a health professional); and
is manufactured using production processes that are capable of being:
either or both validated and verified; and
reproduced.
Most of the devices that were previously supplied under the custom-made medical device exemption will now meet the definition of a patient-matched medical device. Patient-matched medical devices must be included in the ARTG before they are imported into, supplied within, or exported from Australia (unless they are exempt, excluded or otherwise approved by the TGA).
Figure 1: Under the Framework, almost all of the medical devices that previously met the definition of ‘custom-made’ will now meet the definition of ‘patient-matched’. Only a small number of devices will continue to meet the definition of custom-made.
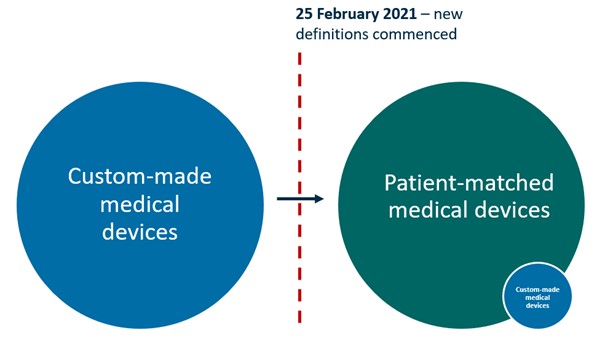
Figure 1. Under the Framework, almost all of the medical devices that previously met the definition of ‘custom-made’ will now meet the definition of ‘patient-matched’. Only a small number of devices will continue to meet the definition of custom-made.
{kind=link}
Illustration showing two large circles connected by an arrow. The left blue circle represents 'Custom-made medical devices'. The right green circle represents 'Patient-matched medical devices', which includes a smaller blue circle labelled 'Custom-made medical devices'. A red dotted line marks February 25 2021, when new definitions commenced. The arrow indicates the transition between these classifications.
Most of the devices that were previously supplied under the custom-made medical device exemption will now meet the definition of a patient-matched medical device (PMMD). To enable the continued manufacture and supply of PMMDs a transition period was established.
If you intend to supply your PMMD on, or after 1 July 2029 you must notify the TGA of the intention to transition before 1 November 2024.
Under the Therapeutic Goods (Medical Devices) Regulations 2002, all PMMDs will continue to be exempt from ARTG inclusion until 1 July 2029 and can be manufactured and/or supplied until this date.
After 1 July 2029, all patient-matched medical devices must be included in the ARTG or have a valid application for inclusion underway.
Inclusion of patient-matched medical devices in the ARTG
The transition period for PMMDs will end on 1 July 2029. After this date all PMMDs (including notified PMMDs) must be included in the ARTG before they are imported into, supplied within, or exported from Australia (unless they are exempt, excluded or otherwise approved by us).
Where patient-matched medical devices are being supplied in volumes of five or less per ‘kind' they are able to be supplied without an inclusion in the ARTG under the low volume exemption for patient-matched medical devices.
Exempt devices are exempt from inclusion in the ARTG, but they are not exempt from regulation. Manufacturers and sponsors of exempt medical devices still need to comply with TGA regulatory obligations for medical devices, including meeting the Essential Principles and advertising requirements.
GMDN terms for patient-matched medical devices
Patient-matched medical devices do not need special GMDN terms and may use the same GMDN terms used for devices that are not patient-matched. The manufacturer must choose the GMDN term that is most appropriate and best describes their device and how it is intended to be used, regardless of whether it is patient-matched or not.
Patient-matched medical devices can use GMDN terms that include ‘custom-made' in the title or description. GMDN terms are internationally agreed generic descriptors used to identify medical devices. The legal definitions for ‘custom-made' and ‘patient-matched' medical devices varies between jurisdictions. The descriptions used in GMDN terms use the phrase ‘custom-made” in line with the ordinary and natural meaning used in everyday language. Generally, you can use the term ‘custom-made' to indicate that a device is designed and manufactured to suit an individual.
For example, the most appropriate GMDN term for a patient-matched orthotic insole might be ‘GMDN 62870 - Orthotic insole, custom-made'. In this instance ‘custom-made' would simply mean a device personalised to meet an individual's specific requirements and the description could cover both patient-matched medical devices and custom-made medical devices.
If the manufacturer cannot find a suitable GMDN term to match the characteristics and intended purpose of their device, the manufacturer can contact the GMDN Agency and request assistance. In some cases, the GMDN Agency will need to create a new GMDN term.
Specified design envelope
The definition of a specified design envelope, as set out in the Regulations, is as follows:
specified design envelope means minimum and maximum dimensions, performance limits or other relevant factors that:
characterise a medical device for production purposes; and
may be based on a standard device template.
The specified design envelope is the limits of design that a manufacturer can be confident (with the support of objective evidence) will result in a medical device that is safe and fit for its intended purpose and will meet the intended recipient's requirements (i.e., design validation).
A design envelope may include several factors, such as:
- minimum and maximum dimensions
- performance limits
- allowable environmental limits for operation
- specifications for materials and their properties.
Factors that inform the specified design envelope could include:
- clinical research
- industry standards
- clinical practice guidelines
- specialist education or training
- on-the-job experience producing and using medical devices.
Example - Specified design envelope
Eleanor is a podiatrist who intends to produce patient-matched therapeutic insoles for her patients. As part of the technical documentation she is compiling through the conformity assessment process, Eleanor establishes and documents a specified design envelope for these devices.
Eleanor considers the following:
- the range of pathologies she knows she can treat using her patient-matched insoles, based on her training and professional experience and supported by knowledge held by her profession more broadly
- what design features she knows are suitable for use in treating each pathology (e.g., forefoot valgus wedge for treating plantar fasciitis)
- what materials she knows she can and cannot use for each kind of insole she produces
- the foot sizes she knows her equipment can and cannot accommodate.
While documenting this information, Eleanor notes that her specified design envelope is quite broad and consequently she is able to accommodate almost any patient who presents to her. This is because her devices and their use in clinical practice are very well-characterised within her industry. As a result, Eleanor is unlikely to ever need to produce a custom-made insole.
Eleanor concludes that her products are all patient-matched medical devices.
Production processes that can be validated and/or verified, and reproduced
Process validation, product verification and reproducibility are key concepts in medical device production.
Process validation refers to establishing, by objective evidence, that a process consistently produces a product meeting predetermined requirement. When used in the medical device context, process validation means a process has been subject to such scrutiny it can be virtually guaranteed to produce devices of a consistent quality. It is important to note that both automated and manual processes can be validated.
Validation is particularly important if the predetermined requirements of the product can only be assured by destructive testing. Factors such as production volume and number of manufacturing steps per unit may influence how process validation is undertaken. Manufacturers can, and should, seek out and select technology-specific guidance and applicable technical standards on applying process validation to their circumstances.
Verification refers to confirmation by objective examination of a product that the predetermined requirements of the product have been met. For example, measuring a device to ensure it has the required dimensions is an example of a verification procedure.
When process validation and product verification are applied, they result in production processes that will consistently produce devices that have similar characteristics, are of a similar quality and perform in a uniformly reliable manner. In other words, the outcome from the process is reproducible. This does not mean that any two devices produced are the same - only that the manufacture of all devices is underpinned by consistent factors that can be justified, ultimately ensuring that a medical device is safe and fit for its intended purpose and will perform as intended.
It is important to note that the manufacture of some medical devices, including those manufactured using additive or subtractive methods require humans to be involved in the production process - providing hand finishing and verification activities, for example. The involvement of a human factor in a manufacturing process does not mean that a manufacturing process cannot be validated and/or verified and reproduced.
Low volume supply of patient-matched medical devices
A low volume exemption exists for manufacturers and sponsors who are importing, exporting or supplying five (5) or less of a ‘kind' of patient-matched medical device in any financial year. While patient-matched medical devices that are supplied in very low quantities do not need to be included in the ARTG before they are imported, exported or supplied, they must meet all other Australian regulatory requirements for medical devices.
For more information, see the guidance about the ongoing responsibilities for sponsors of a medical device.
Example - Low volume exemption
Shikha is an optometrist who sources contact lenses from an Australian sponsor for use in her practice. Some of Shikha's clients have a low tolerance for wearing contact lenses because their eyes become irritated quite quickly. Recently Shikha was contacted by CuCme, an overseas manufacturer of hard contact lenses that are designed and manufactured for specific individuals using a high-resolution scan of the patient's eye as the basis for a contact lens design.
CuCme claim their contact lenses are more comfortable because of their unique approach to design and manufacture. While they use the phrase “custom-made” in their advertising material, the lenses are made using consistent materials and methods that can be validated, verified and reproduced. The personalisation of the device using the scan of the patient's eye does not mean the device meets the definition of custom-made but is in fact patient-matched.
Because they are hard contact lenses, only one pair per year are required per patient. Under the low volume exemption for patient-matched medical devices, Shikha can import and supply up to five patient-matched contact lenses from CuCme per financial year without including the lenses in the ARTG.
If Shikha decides that she would like to import more than five a year for her patients, Shikha will need to ensure that the contact lenses have been included in the ARTG before they are imported. Shikha may take responsibility for this herself so she can import them directly, or she may use an Australian-based sponsor who has included the devices in the ARTG and is prepared to import and supply them to her.
Shikha must meet all other regulatory requirements for medical devices.
More information
Other resources that may assist you to better understand these concepts, include:
- the Global Harmonisation Task Force (GHTF) document Quality Management Systems - Process Validation; and
- ISO 13485:2016 Medical devices - Quality management systems - Requirements for regulatory purposes.
- The International Medical Device Regulators Forum Personalized Medical Devices Working Group has produced three key documents that may assist you:
Example - Patient-matched medical device
Dean's company manufactures mass-produced orthopaedic implants but has also developed a capability to personalise certain devices in their catalogue where necessary. Dean is contacted by an orthopaedic surgeon who needs a personalised acetabular cage and cup manufactured for Dorcas, an 81-year-old female patient who needs to undergo a revision procedure complicated by complete loss of the anterior column and marked bone loss through the remaining acetabulum.
Dean reviews the information sent through by the surgeon including Dorcas' age, height, and weight, and determines that the design and production considerations for her device fall within the scope of what his company knows it can safely produce (i.e., their design envelope). The surgeon sends through CT imaging data to help inform the design of the device and consults with Dean's company on certain features such as how the device should attach to the bone. Dean employs the same production and verification methods to produce Dorcas' implant as he has for dozens of other personalised devices.
In this example, the device meets the definition of a patient-matched medical device because it:
- has been designed by the manufacturer within a specified design envelope to fit the particular anatomy and physiology of a particular individual; and
- has been produced using a process capable of being validated and/or verified and reproduced.
Counter example
Dean is contacted again by the orthopaedic surgeon, this time to manufacture an acetabular for Jake. Jake is a 43-year-old male patient who is 2.26 metres tall and weighs 160 kilos. Dean has never made an implant for someone so tall or heavy, and Dean determines that the dimensions and tolerances of the implant required for Jake fall well outside the design envelope that his company has validated. The surgeon has spoken to several producers and finds that Jake's requirements are not catered to by the design envelope of any patient-matched acetabular cage.
Dean uses the technical and design files to inform a modified device to meet Jake's requirements. He uses computer modelling to perform an engineering assessment to ensure the device he is producing will withstand the forces reasonably expected to be exerted during normal use but does not have the capacity to conduct a full clinical assessment or evaluation for what will be a one-off design. Dean communicates with Jake's surgeon, who uses his expertise and clinical judgement to inform the design of the device. Jake is made aware that his device is truly unique and informed about the risks associated with its use.
Dean employs the same production and verification methods to produce Jake's implant as he has dozens of other personalised devices.
The resultant device meets the definition of a custom-made medical device because the device:
- is intended for the sole use of the intended recipient (Jake)
- has been made at the request of a healthcare professional (the orthopaedic surgeon)
- who has determined that there are no alternative devices available on the ARTG to address the specific needs of this patient to an appropriate level
- does not meet the definition of a patient-matched or adaptable medical device.
In this case, while the manufacturing process can (and has) been validated, the device is being produced outside the design envelope. The resultant product therefore meets the definition of a custom-made device and will continue to be exempt from inclusion in the ARTG subject to the conditions outlined previously.
Adaptable medical devices
The definition of an adaptable medical device, as set out in the Regulations, is as follows:
adaptable medical device means a mass-produced medical device that is intended by the manufacturer to be assembled or adapted after it has been supplied, in accordance with the manufacturer's instructions, to:
address either or both of the anatomical and physiological features of a particular individual; or
address a pathological condition of a particular individual; or
otherwise perform as intended by the manufacturer.
The definition has been included in the Regulations for the sake of clarity but there is no change to the way devices that meet this definition are regulated. Adaptable medical devices continue to be regulated through inclusion in the ARTG under an appropriate classification.
Information to be supplied with an adaptable medical device
Essential Principle 13.4(3 - item 30) applies to adaptable medical devices and requires manufacturers of these devices to ensure that any adaptable medical device that they produce is supplied with instructions for assembling or adapting the device that, if followed by intended users, will ensure the device continues to comply with all relevant Essential Principles.
Example - Adaptable medical device
Tamara's company supplies a mass-produced polymer surgical implant for cranial reconstruction that is:
- supplied in a sterile state
- intended to be thermoformed during the cranial reconstruction procedure to suit the individual patient's anatomical features.
Tamara's implant meets the definition of an adaptable medical device because it is:
- mass-produced
- intended by the manufacturer to be assembled or adapted after it has been supplied to address an anatomical feature of the intended recipient.
Under the new personalised medical devices framework this product will continue to require an inclusion in the ARTG before it can be supplied. It will also need to be supplied with instructions for use that will allow the surgeon using it to safely heat and shape the polymer to suit the patient's anatomy, ensuring the device continues to be safe and fit for its intended purpose after it has been adapted.
Counter example - not a manufacturer
Brett is a rehabilitation engineer. During a consultation, a patient who wears an off-the-shelf ankle-foot orthosis (AFO) asks for Brett's help as she is finding the AFO painful to wear. Brett reviews the AFO, and determines some minor adjustments can be made, such as the addition of some padding, to address this issue for the patient.
Brett wants to understand if he will become the manufacturer of an AFO if he makes these adjustments.
Brett reviews the information available on the TGA website and notes that under section 41BG(3) of the Act, a person does not meet the definition of a manufacturer if:
the person assembles or adapts the device for an individual patient
the device has already been supplied by another person
the assembly or adaptation does not change the purpose intended for the device by means of information supplied by that other person, on or in any one or more of the following:
the labelling on the device
the instructions for using the device
any advertising material relating to the device
technical documentation describing the mechanism of action of the device.
Brett determines that the modifications he has made, while not necessarily intended by the manufacturer of the AFO, do not make him the manufacturer because the modifications do not change any of the features listed under (c) above.
Custom-made medical devices
The below is a summary only.
Detailed information about the regulatory requirements that specifically apply to custom-made medical devices is available on the TGA website: Custom-made medical devices: Information for sponsors, health professionals & manufacturers.
A custom-made medical device statement template has been included at Appendix 2 to help manufacturers and sponsors of medical devices meet their regulatory requirements for custom-made medical devices.
The definition of a custom-made medical device, as set out in the Regulations, is as follows:
custom made medical device means a medical device that:
is intended by the manufacturer to be for:
the sole use of a particular patient (the intended recipient); or
the sole use of a particular health professional (the intended recipient) in the course of the health professional's practice; and
is manufactured by the manufacturer in accordance with a written request of a health professional (the requesting health professional) and with particular design characteristics specified by that health professional in the request (even if the design is developed in consultation with the manufacturer), where those design characteristics are intended to address:
either or both of the anatomical and physiological features of the intended recipient; or
a pathological condition of the intended recipient; and
the requesting health professional has determined is necessary to address the matters covered by paragraph (b) because there is no kind of medical device included in the Register to address those matters or to address those matters to an appropriate level.
However, a custom-made medical device does not include a patient matched medical device, an adaptable medical device or other mass-produced medical device.
Custom-made or patient-matched
Both custom-made and patient-matched devices are designed and produced for a particular individual. In addition, both custom-made and patient-matched medical devices can be manufactured in accordance with a written request from a health professional. The key difference between these definitions is that a custom-made device is so rare and unique that there is no way that the manufacturer can adequately validate the design of the device, or adequately validate and/or verify the production process, at the time it is requested.
Custom-made medical devices can only be produced and supplied where a patient's particular circumstances mean that there is no other suitable device available in the ARTG for use in their treatment.
If you manufacture a medical device in accordance with the written request of a health professional to suit an intended recipient, it does NOT mean that your device is custom-made.
Both custom-made and patient-matched medical devices can be manufactured in accordance with the written request of a health professional. This is not a distinguishing feature for custom-made medical devices.
| Type of personalised medical device | Designed and produced for the sole use of a particular individual? | Routinely-made by the manufacturer? | Is it feasible to collect objective evidence of the safety and performance of the device? |
|---|---|---|---|
| Custom-made | Yes | No | No |
| Patient-matched | Yes | Yes | Yes |
Wherever possible, patients and health professionals should access medical devices that are ARTG-included and supported by appropriate evidence of their quality, safety and performance. The custom-made medical devices exemption addresses the rare circumstances where the rarity of the clinical presentation and needs of the end-user limit the availability of evidence.
When a device is exempt from inclusion in the ARTG, it does not mean the device is exempt from TGA regulation entirely. Manufacturers and sponsors of custom-made medical devices still have regulatory obligations that they must meet under TGA legislation.
Example - Custom-made medical device for a patient
Samara is an orthopaedic surgeon. Samara is asked to review a patient who presented to the emergency department with loss of elbow function and severe pain following a traumatic fall. CT imaging demonstrates significant injury to the radial head, with loss of viable bone throughout the proximal portion of the radius. Samara determines the radial head will need to be replaced to restore functionality to the elbow; however, the loss of bone in the proximal portion of the radius is significant.
Samara determines there is no device included in the ARTG that could be used to reconstruct the radial bone. She approaches a manufacturer of orthopaedic devices to request they produce a proximal replacement radius for the patient, making use of the patient's own bone to form part of both the matrix and reinforcement component of the replacement implant. Samara uses her knowledge and experience as an orthopaedic surgeon to determine key design characteristics (including the angle of the radial head to the stem and the composition of materials) that should provide the best outcome for the patient.
Samara provides these characteristics and the patient's CT scanning data to the manufacturer to help inform part of the design. The manufacturer designs and produces a proximal radial implant for the patient based on the information supplied by Samara.
In this example, the proximal radial implant is a custom-made medical device because the device:
- is intended for the sole use of the intended recipient
- is designed with particular design characteristics (e.g., the angle of the radial head to the stem) specified by a health professional (Samara) to address the anatomical features of the intended recipient
- was manufactured because there were no alternative devices available on the ARTG to address the intended recipient's needs to an appropriate level, owing to the degree of injury and its rarity
- does not meet the definition of a patient-matched medical device because although the manufacturer has experience with these types of device, the degree of bone loss means they have had to design this device well outside the parameters of their specified design envelope
- does not meet the definition of an adaptable medical device because the device is not mass produced, and is not intended to be adapted after supply.
Example - custom-made medical device for a health professional
Tony is a gastroenterologist who has lost some dexterity because of nerve damage sustained during an accident. He employs a biomedical engineer to design and manufacture a modified steering mechanism for an endoscope to help him manage the loss of dexterity, thereby allowing him to continue operating the endoscope safely. Tony dictates the design characteristics that he will need for the steering mechanism including the level of responsiveness needed.
The engineer devises a solution to fit. In this example, the steering mechanism will meet the definition of a custom-made medical device because the device:
- is intended for the sole use of the intended recipient (the gastroenterologist);
- is designed by a health professional (Tony) to address the physiological features of the intended recipient;
- was manufactured because there were no alternative devices available in the ARTG to address the intended recipient's needs;
- does not meet the definition of a patient-matched medical device because it has been produced outside of the manufacturer's specified design envelope as a one-off for Tony; and
- does not meet the definition of an adaptable medical device because it is not being mass-produced, and it is not intended to be adapted after supply.
Counter example - not a custom-made medical device
Sharni's company produces personalised maxillofacial plates that can be manufactured to suit a patient's unique anatomy. DICOM files are sent to Sharni by each referring surgeon. Sharni imports the DICOM file data to her computer-aided design and manufacture (CAD/CAM) program and designs a plate to fit the patient's specific defect.
Sharni confirms the design with each requesting surgeon before commencing production of the final device.
Sharni's maxillofacial plates will not meet the definition of a custom-made medical device because they are manufactured:
- within a specified design envelope; and
- using production processes that can be validated and/or verified, and reproduced.
Sharni's plates meet the definition of a patient-matched medical device. Sharni will need to make transition notifications for the devices by 1 November 2024 if she intends to supply them after 1 July 2029.
Medical device production systems (MDPS)
Medical Device Production Systems (MDPS) are a new regulatory concept, designed to provide options to healthcare facilities wanting to produce patient-matched device in-house for treating their patients.
The TGA is currently working with a number of stakeholders, including the IMDRF, to ensure appropriate procedures and processes are in place to enable the safe introduction of this new concept.
For this reason, this facility is not currently available and an MDPS cannot be approved by the TGA or included in the ARTG at this time.
If you are a health professional currently using a system to produce patient-matched medical devices, you currently meet the definition of a manufacturer under section41BG of the Act. You must ensure you are meeting all of the relevant regulatory obligations.
If you would like to be notified when MDPS can be approved, subscribe to the Personalised Medical Devices subscriber list, by sending an email to devices@tga.gov.au with “SUBSCRIBE PMD” in the subject line.
The definition of a medical device production system (MDPS), as set out in the Regulations, is as follows:
medical device production system means a system that consists of raw materials and main production equipment (whether or not the system also consists of software), where the system is intended by the manufacturer to be used (whether or not with ancillary inputs or equipment) by a health professional, or suitably qualified person within a healthcare facility, to produce a particular medical device for use in relation to a patient of the health professional or healthcare facility.
An MDPS is a validated, multi-component design and production system intended to produce a patient-matched medical device within a healthcare facility.
Figure 2. A Medical Device Production System (MDPS) consists of all the required components to produce a patient-matched medical device, from raw materials to production equipment.
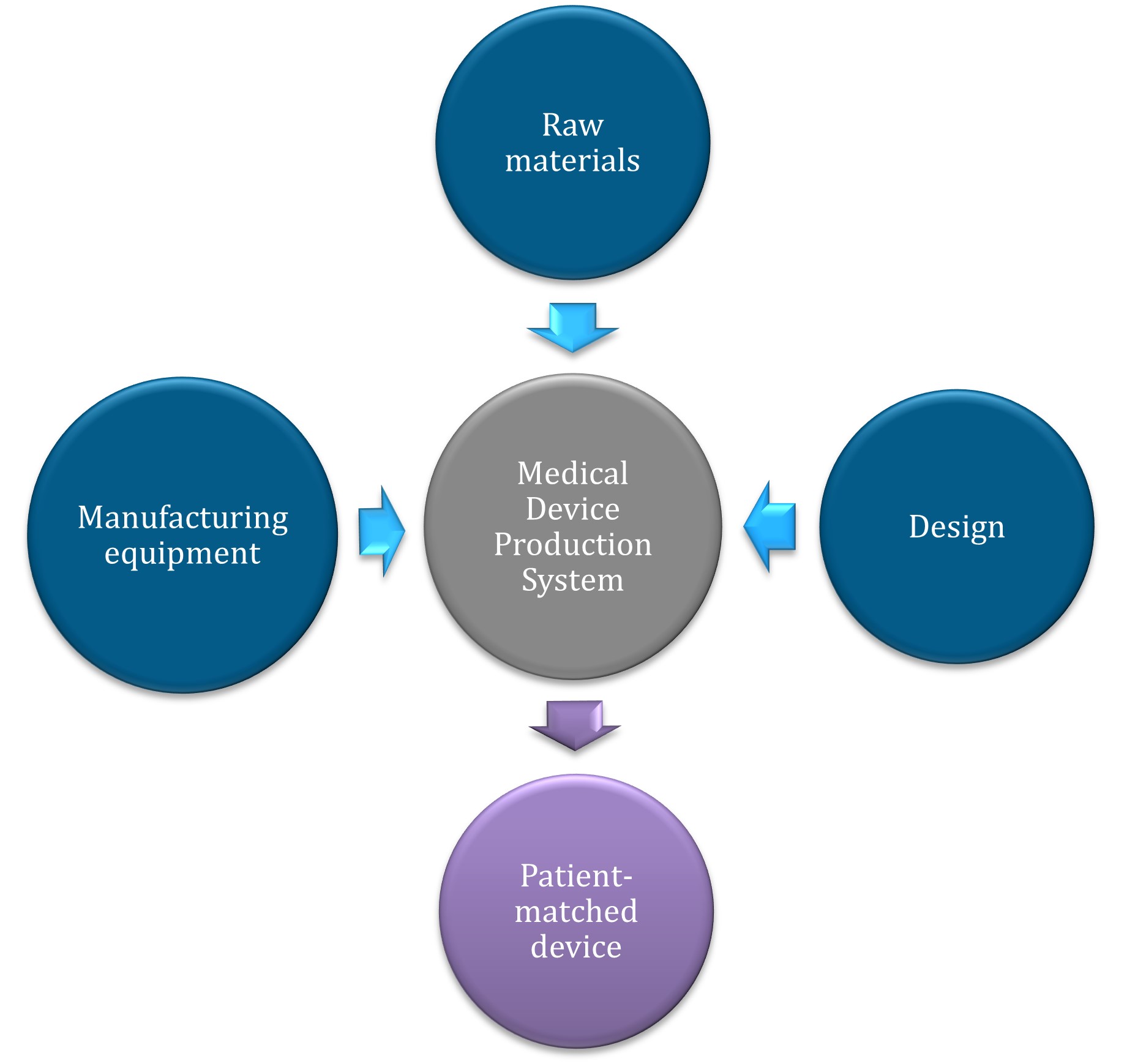
Figure 2. A Medical Device Production System (MDPS) consists of all the required components
to produce a patient-matched medical device, from raw materials to production equipment.
{kind=link}
A vertical flow diagram using circular nodes and arrows to show a medical device production system.
The diagram has 3 input nodes at the top and sides:
- raw materials (top, dark blue)
- manufacturing equipment (left, dark blue)
- design (right, dark blue).
These 3 inputs connect via light blue arrows to a central grey node labelled 'Medical Device Production System'.
From this central node, a purple arrow points downward to the final output node in purple, labelled 'Patient-matched device'.
The layout emphasises a streamlined workflow from multiple inputs to a single customised output. All text is displayed in white within each circular node, and the nodes have a subtle shadow effect.
Where a manufacturer includes an MDPS in the ARTG, the conformity assessment process for the MDPS will include the resultant devices. When a healthcare professional or suitably qualified person within a healthcare facility within a healthcare facility uses an MDPS that has been included in the ARTG to manufacture a patient-matched medical device, they will not be required to:
- undergo a conformity assessment for the patient-matched device they are manufacturing; or
- include the device they are manufacturing in the ARTG.
Users of MDPS
Once the MDPS facility is available, if you are a health professional or suitably qualified person within a healthcare facility who produces patient-matched medical devices, you will be able to supply devices providing:
- you purchase and use an MDPS that has been included in the ARTG
- you only use the MDPS to produce devices in accordance with the intended purpose and instructions for use as stated by the manufacturer.
If you:
- manufacture medical devices from a production system not included in the ARTG as an MDPS; or
- use an ARTG-included MDPS to produce a medical device other than the kind of medical device intended by the manufacturer of the MDPS
then you will need to meet all regulatory requirements associated with the manufacture and supply of a medical device.
By using an ARTG-included MDPS in accordance with the manufacturer's instructions, you will not need to:
- apply the relevant conformity assessment procedure yourself to the patient-matched devices produced by the MDPS; or
- include the patient-matched medical devices produced by the MDPS in the ARTG before you supply them to a patient.
Example Medical Device Production System (MDPS)
Kate has developed a ceramic milling system that she intends to include in the ARTG and supply to dentists, who can use the system to produce patient-matched dental crowns for adult patients. These crowns are to be used with standard dental implants and abutments from a line that Kate's company also supplies. The system incorporates:
- ceramic blocks
- a ceramic milling machine
- a furnace for firing
- post-machining finishing equipment
- CAD/CAM proprietary software that:
- reads files generated from intraoral scans
- designs a patient-matched dental crown for a particular patient according to the scans
- controls the production equipment.
Kate intends to restrict use of the MDPS to dentists, dental nurses and dental technicians in accredited practices that have attended a training course in how to safely use and maintain the MDPS.
This system meets the definition of an MDPS because:
- Kate has specifically intended for it to be an MDPS
- it includes raw materials and main production equipment intended to be used together as a system
- the system is intended by the manufacturer to be used by dentists in their capacity as health professionals
- the system is intended to produce personalised medical devices for patients of the dental clinic.
Example Using an MDPS to produce a device other than intended
Achara is a dentist who wants to buy an MDPS from Kate for use in her practice. Achara has previously used an overseas supplier to procure zirconia blocks for far cheaper than what Kate is offering to sell them for as part of the MDPS. Achara wants to know if she can swap out Kate's blocks for the ones she imports herself.
If Achara uses materials other than those supplied by Kate's company, she will meet the definition of a manufacturer because she is not using the MDPS in accordance with Kate's Instructions for Use, as validated through the conformity assessment process.
Kate's company cannot account for the composition of non-genuine materials, and so cannot accept responsibility for ensuring that any devices produced using it are safe and fit for their intended purpose. Given also that the crown is not used in isolation but as part of a system with the dental implant and abutment, the use of non-genuine materials would mean that Achara becomes the legal manufacturer of these components as well. Achara would therefore become the legal manufacturer of all three of these components, and would need to meet all of the relevant regulatory obligations as the manufacturer of a medical device, including undergoing an appropriate conformity assessment certification process through a third party (noting that a dental implant is a Class IIb medical device at minimum).
Counter example - A production system that is not an MDPS
A team of medical physicists working in a hospital radiation oncology department have put together a system for producing 3D-printed, patient-matched (medical device) boluses in-house for their patients. The physicists have put the system together themselves, selecting raw materials, design and production equipment based on their expertise. The team does not plan to commercialise its system to sell on to other hospitals.
- the team members are not using an MDPS because the system has not been supplied to them by a third party as a complete system, they have put it together themselves
- the physicists are not the manufacturers of an MDPS because they do not intend to supply the system.
The physicists are manufacturers of patient-matched medical devices and will need to submit an application for inclusion of their boluses in the ARTG.
Who is a health professional or a suitably qualified person within a healthcare facility?
The Regulations define a health professional as:
health professional includes a person who is:
a medical practitioner, a dentist or any other kind of health care worker registered under a law of a State or Territory; or
a biomedical engineer, chiropractor, optometrist, orthodontist, osteopath, pharmacist, physiotherapist, podiatrist, prosthetist or rehabilitation engineer.
It is recognised that professionals not listed in the definition above may also work in healthcare facilities and:
- provide healthcare services; or
- support health professionals to deliver healthcare services.
This includes, but is not limited to:
- allied health assistants
- speech pathologists
- medical physicists
- clinical scientists
- orthotists
- occupational therapists
- medical and dental laboratory technicians or technical officers
- prosthetic subspecialties (e.g. ocularists, anaplastologists)
- dental technicians.
The degree to which any user is 'suitably qualified' to operate an MDPS will depend upon the device to be produced, the complexity and needs of the system, and the training and experience held by, or available, to them. The manufacturer of an MDPS will be required to state in the instructions for use the requirements they have of a 'suitably qualified' user. This could include minimum qualifications and experience, job title or classification, accreditation by a third party and/or attendance at training courses run by the manufacturer.
If the manufacturer's explicit instructions regarding who should be operating the MDPS are not followed, then the MDPS is not being used in accordance with the manufacturer's instructions. As a result, the user of the system would be responsible for all relevant regulatory obligations as a manufacturer and sponsor of a patient-matched medical device.
Supplying devices without an ARTG inclusion is a breach of the Act. Civil and criminal penalties apply.
Manufacturers and sponsors of an MDPS
If you are the manufacturer or supplier of a Medical Device Production System (MDPS), you will need to ensure that:
- the appropriate conformity assessment procedures have been applied to the MDPS relevant to its classification, demonstrating it, and all devices it produces, meet all relevant Essential Principles
- the MDPS is included in the ARTG with the same classification as the highest class of device it is intended to produce.
The TGA recognises that the concept of an MDPS is new, and that manufacturers and sponsors may need assistance to ensure they comply with the Regulations. You may wish to contact PersonalisedDevices@health.gov.au to discuss your specific circumstances.
Example Medical device production system (MDPS)
Zane leads a research team that has developed a system for the production of patient-matched, 3D-printed splints for use in immobilising patients' arms while broken bones heal. Zane intends to commercialise the system and sell it to hospitals as an MDPS, allowing orthopaedic departments to produce patient-matched 3D-printed splints in-house.
The system is comprised of some components manufactured by Zane and some sourced from other manufacturers. The system consists of:
- CAD/CAM software that provides instructions to the system
- a 3D printer
- raw materials
- tools for finishing the devices produced (e.g., sandpaper for sanding down support struts leftover from the printing process).
Before they can apply to the TGA to have their MDPS approved and included in the ARTG, Zane and his team will need to apply the appropriate conformity assessment procedures and ensure that all of the relevant Essential Principles are met both for the MDPS and the devices it produces.
Zane will need to take into consideration a number of factors including:
- Who is the manufacturer? Even though Zane's team only produces certain components of the system, they will be the legal manufacturer of the MDPS under section 41BG(2) of the Act because they will be responsible for assigning to the system its purpose through labelling, Instructions for Use, advertising material and technical documentation.
Zane will need to review the information about manufacturing medical devices available on the TGA website and ensure he understands the obligations he and his team will now have. He will need to manage third-party suppliers (including the producers of the components) under his quality management system to ensure they continue to provide components / raw materials of sufficient specified quality, as well as holding, and being able to provide him with, suitable documentation as required by the Regulations.
- What must be supplied as part of the system, and what can be recommended? The splint functions best when it is sufficiently rigid so as to immobilise the fracture site, but not so stiff that it will crack when reasonably anticipated force is exerted on it (for example, if the patient were to fall or hit the splint on a door accidentally). Zane determines that the performance of the device is heavily dependent on the properties of the raw material from which it is produced. Zane selects a supplier to work with who can provide ongoing assurance that the raw materials supplied are of sufficient and consistent quality to be used in the production of the splints produced by the MDPS.
Zane will supply the system to his customers with the raw material from this specific supplier, and will clearly state in the system's Instructions for Use that only the specific material from the specific supplier he specifies is to be used. If one of Zane's customers sources the same material from a different supplier and uses it to produce devices, they will be operating the system outside of the Instructions for Use and it is the user, and not Zane, who will assume all regulatory responsibility for the devices that are manufactured.
Unlike the raw materials, the sandpaper for finishing the devices does not have any characteristics integral to the safety or performance of the end device. Zane's team can choose to supply sandpaper with the system, or recommend a grade of sandpaper suitable for this purpose and allow the user of the MDPS to source their own sandpaper.
- Who is a suitable user? Zane and his team consider knowledge and skills necessary to both operate and maintain the MDPS to ensure it, and the devices it produces, continue to conform to the Essential Principles once the MDPS has been supplied. Zane and his team mandate minimum qualifications for the users of the MDPS, and put together a two-week training course that must be completed before the MDPS is used. Zane and his team enforce the user requirements by providing those who complete the training course with a unique passcode they must input into the MDPS before they use it. If the individualised passcode is not used, the MDPS will not start.
- How will the quality assurance procedures for the system be carried out and ensured? If the MDPS is not checked and calibrated at specified intervals, Zane and his team cannot guarantee the MDPS will continue to conform to the Essential Principles. In addition to the training course, Zane and his team decide they will automate as much of the quality assurance and quality control (QA/QC) processes as possible.
- They also plan to install safeguards into the system such as:
- reminder notices to perform routine QA/QC tasks
- an automated feature that will shut down the MDPS if QA/QC is not carried out, is carried out inappropriately or if the pre-imposed limits are exceeded.
Zane and his team will also commit to performing routine check-ins with their customers by visiting them on-site, using these visits to perform manufacturer-specific maintenance activities and to ensure trained users continue to use the MDPS as intended.
Information to be supplied with an MDPS
Essential Principle 13.4(3) includes a specific requirement for information to be supplied with an MDPS. Manufacturers of MDPSs must ensure they supply their systems with instructions to allow the end-user to produce a medical device that meets the Essential Principles.
These instructions should contain an explicit statement notifying the health professional or suitably qualified person within a healthcare facility using the MDPS that failure to follow the instructions for the system:
- could result in a device not meeting the Essential Principles, which means it may not be safe and fit for its intended purpose
- means they will be responsible for meeting all relevant regulatory obligations for manufacturers and sponsors of patient-matched medical devices.
If an individual:
- produces a device not included in the manufacturer's intended purpose for the system; or
- modifies, changes or adapts the system outside the manufacturer's Instructions for Use; or
- fails to follow the manufacturer's Instructions for Use when using the system;
the individual will be responsible for meeting all relevant regulatory requirements associated with being a manufacturer and sponsor.
What you need to do
If you are the manufacturer or sponsor of a system that you intend to market in Australia as an MDPS, and you would like to be notified when MDPS can be approved, subscribe to the Personalised Medical Devices subscriber list, by sending an email to devices@tga.gov.au with "SUBSCRIBE PMD" in the subject line.
Appendix 1: Personalised medical devices decision tree
Appendix 1: Personalised medical devices decision tree
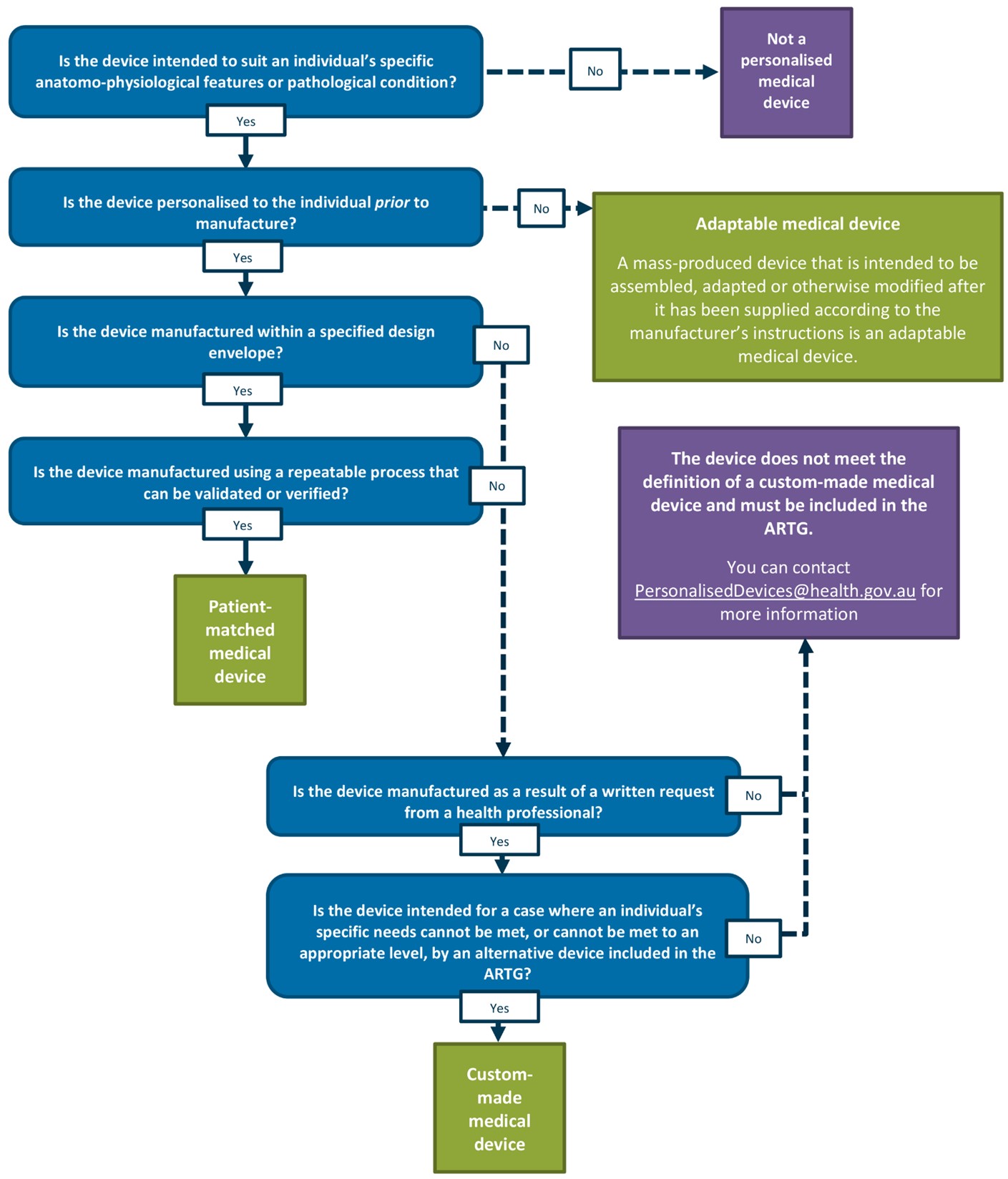
{kind=link}
A vertical decision flowchart using blue question boxes and green/purple outcome boxes to classify medical devices. The flowchart starts with the question 'Is the device intended to suit an individual's specific anatomo-physiological features or pathological condition?' and branches through several decision points:
Top path (No): Leads to 'Not a personalised medical device' (purple box)
Main path (Yes) continues through questions about:
- Pre-manufacture personalisation
- Specified design envelope
- Repeatable/verifiable manufacturing process.
Side branches lead to:
- 'Adaptable medical device' (green box) with definition for mass-produced adaptable devices
- 'Patient-matched medical device' (green box)
- Final section asks about health professional written requests and specific needs
- Ends with either 'Custom-made medical device' (green box) or a purple box explaining non-qualification with ARTG contact information.
The flowchart uses solid arrows for 'Yes' paths and dashed arrows for 'No' paths. Each decision point is clearly marked with Yes/No options in white boxes.
Appendix 2: Statement template for custom-made medical devices
This statement is being supplied with a custom-made medical device in accordance with subclause 7.2(3A) of Schedule 3 of the Therapeutic Goods (Medical Devices) Regulations 2002 (the Regulations).
This custom-made medical device was manufactured by [insert name and legal address of manufacturer]. The device is a [insert a brief description of the device, e.g., transtibial prosthetic sleeve] that can be identified by the following features-
- Briefly outline any identifying features of the device e.g. any branding it may carry, the colour of the material, the size of the device etc.
The device is packaged alone/along with the following-
- List all other contents of the packaging
The device was custom-made for- and intended only to be used in relation to- [insert the name of the individual to whom the device is intended to be used], according to specifications provided by [insert the name and business address of the health professional who provided the specifications for the device].
The following design and/or construction characteristics of the device were specified by [insert the name of health professional who provided the specifications for the device] when they requested the device be manufactured:
| Characteristic | Specifications |
|---|---|
| E.g. length | 15mm |
[Insert the name of the manufacturer] certifies that the device complies/does not comply with the applicable provisions of the Essential Principles of Schedule 1 of the Regulations.
If the device does not comply with any of the applicable provisions of the Essential Principles:
The device does not comply with essential principle/s [insert the numbers of the applicable Essential Principles that the device does not conform to] because [insert the reason for non-compliance].
This statement will be kept on file by [insert the name of the manufacturer] for 5 years [if the device is non-implantable]/15 years [if the device is implantable], in accordance with subclause 7.6(2) of Schedule 3 of the Regulations.
This statement was compiled by the person named below, in accordance with the requirements of subclause 7.2(2) of Schedule 3 the Regulations.

Page history
Updated transition content
Updated to reflect the extension of the notification period for transitioning devices and ARTG inclusion.
Increase readability, fix broken web links, and include additional information on the use of GMDN terms.
Minor update to correct the low volume supply exemption and remove the transition notification deadline date for patient-matched medical devices
Updated to reflect regulation changes under the Therapeutic Goods Legislation Amendment (2021 Measures No. 3) Regulations 2021
Updated to reflect currency of the Framework after the close of the registration period for transitioning devices
Updated to reflect commencement of the Framework and updates to the legislation
Updated to fix formatting issues
Original publication
Updated transition content
Updated to reflect the extension of the notification period for transitioning devices and ARTG inclusion.
Increase readability, fix broken web links, and include additional information on the use of GMDN terms.
Minor update to correct the low volume supply exemption and remove the transition notification deadline date for patient-matched medical devices
Updated to reflect regulation changes under the Therapeutic Goods Legislation Amendment (2021 Measures No. 3) Regulations 2021
Updated to reflect currency of the Framework after the close of the registration period for transitioning devices
Updated to reflect commencement of the Framework and updates to the legislation
Updated to fix formatting issues
Original publication