Summary report of residual DNA and endotoxin on COVID-19 mRNA vaccines conducted by TGA Laboratories
Executive summary
The Therapeutic Goods Administration (TGA) has conducted a comprehensive evaluation of the residual DNA and endotoxin levels in the two mRNA vaccines supplied in Australia - Comirnaty (Pfizer) and Spikevax (Moderna). This report provides scientific evidence confirming that residual DNA and endotoxin levels meet internationally agreed limits.
The TGA's batch release process for COVID-19 vaccines, which involves thorough review and independent laboratory testing, ensures the quality and safety of vaccines before they reach the public. As of 24 October 2024, 212 batches of Comirnaty and Spikevax have been released by the TGA Laboratories. The report includes the results of independent testing by the TGA Laboratories for residual DNA and endotoxins.
Key findings include:
- Residual DNA Testing: The TGA Laboratories tested 28 batches of mRNA vaccines and confirmed that all batches complied with the World Health Organization recommended limit of up to 10 ng per dose. This confirmed the manufacturing process removes almost all DNA starting material and the vaccines are within the safe DNA residual limit. The quantitative PCR (qPCR) method was validated to ensure accuracy and reliability.
- Endotoxin Testing: Every batch of both mRNA vaccines released in Australia was tested for bacterial endotoxins using a validated Limulus amebocyte lysate (LAL) assay. Endotoxin was tested to a level of 5 Endotoxin Units (EU) per mL and all batches were below this level.
- The report concludes that the current mRNA manufacturing processes, as evaluated and approved by the TGA, effectively control levels of residual DNA and bacterial endotoxins to a safe level.
Background
The World Health Organization (WHO) considers batch release as a critical process in ensuring the quality and safety of vaccines[1]. This process involves the evaluation of each individual batch (or lot) of a licensed vaccine before it is released onto the market. This process is typically carried out by National Control Laboratories (NCLs). The evaluation includes a thorough review of the manufacturer’s production data and quality control test results. According to the WHO guidelines, testing is an additional component.
The purpose of batch release is to confirm each batch meets the approved release specifications before it reaches the public. In Australia, batch release of all vaccines is conducted by the TGA Laboratories, which is Australia’s National Control Laboratory for therapeutic goods[2]. The TGA has approved two mRNA vaccines for Severe Acute Respiratory Syndrome Coronavirus 2, (SARS-CoV-2) in Australia. These two vaccines, Comirnaty (Pfizer) and Spikevax (Moderna), constitute the majority of SARS-CoV-2 virus vaccines currently supplied in Australia. Testing was established for these products to ensure critical quality attributes were independently verified by the TGA.
As of 24 October 2024, 212 batches of Comirnaty (154 batches) and Spikevax (58 batches) have been batch released in Australia by the TGA Laboratories. Summary results for all batches are published on the TGA website.
This report contains the more detailed results of the TGA Laboratories independent testing of the two mRNA vaccines for two manufacturing impurities.
- Residual DNA testing may be conducted by the manufacturer prior to final formulation. The TGA Laboratories has reviewed residual DNA levels in the manufacturer’s batch release information or through the evaluation process to ensure it complies with approved specifications. To date, the TGA has also tested 28 batches to confirm the accuracy of the batch data submitted by the manufacturers.
- Endotoxin testing has been included in the batch release testing of all batches of both mRNA vaccines released to date.
Residual DNA
Monitoring for Residual DNA content
Residual DNA may be an impurity in biological medicines that could be present in finished products. It was first encountered as an impurity originating from mammalian cell culture lines used to produce recombinant proteins. It is made up of DNA fragments originating from the host cell used to produce biotechnology medicines or, in the case of mRNA products, the template plasmid used to make the mRNA. Manufacturing controls, such as breakdown through enzymatic DNA digestion and then purification, have been developed and optimised by manufacturers to remove DNA from these preparations. This is evaluated by the TGA prior to marketing authorisation. Safe residual DNA limits, of up to 10 ng per dose, were established for these products to ensure extremely high safety margins[3][4].
Testing for residual DNA may be conducted by the manufacturer at various stages in the manufacture of vaccines and other biological medicines. Acceptable results for these tests provide an assurance that the manufacturing process removes almost all DNA starting material, such that residual levels are below those internationally accepted by the scientific community.
Following the establishment of a manufacturing process which consistently produces comparable commercial batches, with residual DNA levels demonstrated to be below international standard levels, the requirement for residual DNA testing may be safely removed from the release testing process. Consideration of removal of the residual DNA testing should be in accordance with the principles of risk-based regulation. Testing should only be removed if the regulatory authority has assessed the data submitted and there is appropriate justification provided.
Although the test for residual DNA might not be included in the batch release process, routine monitoring may still occur. The TGA Laboratories have independently tested this manufacturing impurity as an additional check. This testing is intended to monitor the effectiveness of the purification process to remove residual DNA.
Residual DNA testing
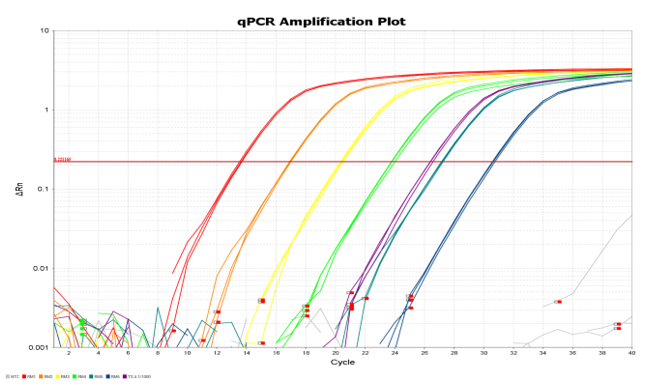
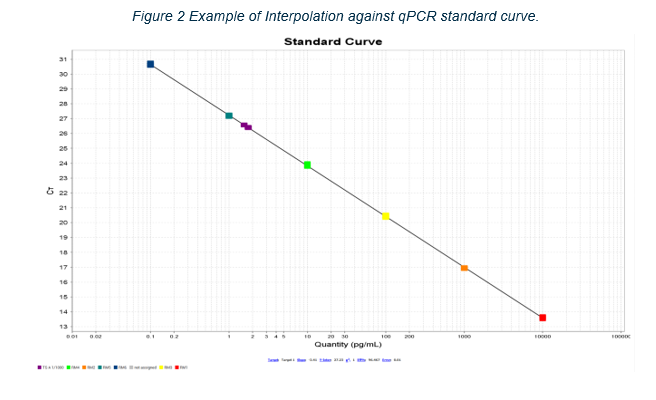
The residual DNA measurements performed by the TGA Laboratories conform to the standard test procedures for residual DNA quantitation available in the European Pharmacopoeia monograph 2.6.35, Quantification and Characterisation of Residual Host-Cell DNA. The test method included in this monograph is quantitative PCR (qPCR). In this test, vaccine samples are compared with standard curves of reference material (plasmid DNA) prepared in a series of known concentrations. The amplification of test samples is compared with that of the standard curve materials to quantitate the amount of residual DNA present in the vaccine, as shown in figures 1 and 2 below. Use of a standard curve and comparison with a measurement from a test sample is a standard method for quantifying substances. The amount of amplification is monitored by a fluorescent reporter, which may either be a dye-labelled DNA probe specific for the gene sequence of interest, or a double stranded DNA-binding fluorescent dye. In the latter case, any background fluorescence arising from RNA is a constant amount that is subtracted from fluorescence measurements in the later cycles during which the exponential amplification curve is observed. Background fluorescence is measured separately for each reaction over the cycles between the green and red markers in the plot below. The cycle at which reaction fluorescence exceeds a certain threshold (where the amplification plots cross the horizontal, maroon line) is the sample threshold cycle (CT) value, and is proportional to the amount of qPCR target DNA initially present in the reaction.
Figure 1 Example of qPCR amplification plot
{kind=link}
Figure 1. Example of qPCR amplification plot. Test samples (purple plots) and standard curve materials (other coloured plots) are amplified by qPCR over multiple cycles of heating and cooling in a thermocycler.
Figure 2 Example of Interpolation against qPCR standard curve
{kind=link}
Figure 2. Example of Interpolation against qPCR standard curve. The CT value for each standard curve reaction is plotted against the amount of qPCR target present in that reaction to create a linear standard curve. The CT values of the test samples (purple markers) are compared with the standard curve to determine the amount of qPCR target DNA present in the test sample reactions.
Endotoxin
Bacterial Endotoxins
High levels of bacterial endotoxins can cause fever if injected into the bloodstream. These endotoxins come from the outer cell membrane of Gram-negative bacteria. Manufacturers of injectable medicines conduct testing to ensure that levels of bacterial endotoxins are minimised as far as possible in the finished product. The TGA evaluates endotoxin testing information from manufacturers of injectable medicines, including vaccines, to ensure these standards are met prior to approval.
Bacterial Endotoxin testing
There are standard methods for conducting bacterial endotoxin testing. These are internationally accepted methods that are harmonised across the European Pharmacopoeia (Ph.Eur. 2.6.14) and United States Pharmacopoeia (USP <85>). These methods are used by manufacturers across the world for testing injectable medicines.
This pharmacopoeial6 testing must be verified following certain procedures to be considered compliant. The TGA uses an endotoxin test that is compliant with these internationally standardised methods.
The method used must demonstrate that endotoxin can be detected without interference from the product itself. The currently registered mRNA vaccines contain lipids. When originally verifying the method, the TGA needed to demonstrate that a known amount of endotoxin spiked7 into the products could then be detected in the assay. Our bacterial endotoxin assay for both the currently registered mRNA vaccines has been fully verified and is compliant with pharmacopoeial requirements8.
Samples tested
Residual DNA testing commenced in late 2023 using samples of all batches available for distribution within Australia at that time. Testing was performed on formulated vaccine samples that were provided to the TGA Laboratories for batch release testing. To date, 28 batches of mRNA vaccines have been independently monitored by the TGA Laboratories for residual DNA content. These samples were shipped and stored under controlled conditions that were continually monitored, to ensure compliance with the instructions for storage and use of the vaccine products. All samples were tested before the expiry date. All batches of both mRNA vaccines were tested for endotoxin.
Testing results
Residual DNA - qPCR
All 28 batches tested at the TGA Laboratories passed the WHO recommended limit for residual DNA of up to 10 ng /dose.
Moderna (Spikevax)
13 batches were tested at the TGA Laboratories, all of which complied with WHO recommended residual DNA limit of <10ng/dose (Table 1).
| Batch | Below WHO recommended limit | Batch Expiry | Residual DNA content (ng/dose) | Expanded uncertainty 95% CI9 | AUST R10 |
|---|---|---|---|---|---|
| 0000039 | COMPLIES | 05-Dec-23 | 0.58 | 0.23 – 0.93 | 399553 |
| 0000040 | COMPLIES | 11-Dec-23 | 0.89 | 0.36 – 1.43 | 399553 |
| 0000041 | COMPLIES | 14-Dec-23 | 0.65 | 0.25 – 1.04 | 399553 |
| 0000042 | COMPLIES | 20-Dec-23 | 0.55 | 0.22 – 0.88 | 399553 |
| 0000037 | COMPLIES | 19-Nov-23 | 0.40 | 0.16 – 0.64 | 399553 |
| 0000038 | COMPLIES | 22-Nov-23 | 0.44 | 0.18 – 0.70 | 399553 |
| 0000036 | COMPLIES | 13-Nov-23 | 0.36 | 0.14 – 0.58 | 399553 |
| 3034111 | COMPLIES | 30-Jun-24 | 0.07 | 0.03 – 0.11 | 418911 |
| 3034112 | COMPLIES | 30-Jun-24 | 0.08 | 0.03 – 0.13 | 418911 |
| 3034113 | COMPLIES | 31-Jul-24 | 0.04 | 0.02 – 0.06 | 418911 |
| 3034347 | COMPLIES | 31-Aug-24 | 0.04 | 0.02 – 0.06 | 418911 |
| 3034346 | COMPLIES | 30-Sep-24 | 0.05 | 0.02 – 0.08 | 418911 |
| 3036548 | COMPLIES | 30-Sep-24 | 0.05 | 0.02 – 0.09 | 418911 |
Pfizer (Comirnaty)
15 batches were tested at the TGA Laboratories, all of which complied with WHO recommended residual DNA limit of <10ng/dose (Table 2).
| Batch | Below WHO recommended limit | Batch Expiry | Residual DNA content (ng/dose) | Expanded uncertainty 95% CI | AUST R |
|---|---|---|---|---|---|
| GL7687 | COMPLIES | 31-Aug-24 | 0.86 | 0.34 – 1.38 | 400874 |
| GL3978 | COMPLIES | 31-Aug-24 | 3.20 | 1.28 – 5.12 | 400874 |
| GK3908 | COMPLIES | 31-Aug-24 | 2.90 | 1.16 – 4.64 | 400874 |
| 2F3007A | COMPLIES | 31-Aug-24 | 0.48 | 0.19 – 0.77 | 400874 |
| GM6363 | COMPLIES | 31-Aug-24 | 0.63 | 0.25 – 1.00 | 400874 |
| GK3907 | COMPLIES | 31-Aug-24 | 2.49 | 1.00 – 3.99 | 400874 |
| GK1316 | COMPLIES | 31-Aug-24 | 0.76 | 0.30 – 1.22 | 400874 |
| 2F3019A | COMPLIES | 31-Aug-24 | 1.41 | 0.56 – 2.25 | 400874 |
| HH1297 | COMPLIES | 31-Jan-25 | 5.56 | 2.22 – 8.90 | 419330 |
| HJ3095 | COMPLIES | 28-Feb-25 | 5.05 | 2.02 – 8.08 | 419330 |
| HG2252 | COMPLIES | 30-Nov-24 | 5.12 | 2.05 – 8.18 | 419330 |
| HL9694 | COMPLIES | 31-Aug-24 | 0.81 | 0.32 – 1.30 | 419371 |
| HJ3101 | COMPLIES | 31-Mar-25 | 5.91 | 2.36 – 9.46 | 419330 |
| HJ3372 | COMPLIES | 28-Feb-25 | 1.20 | 0.48 – 1.92 | 419330 |
| LD8974 | COMPLIES | 30-Sep-25 | 0.05 | 0.02 - 0.08 | 419332 |
Endotoxin – LAL (Limulus amebocyte lysate)
This assay is used to detect bacterial endotoxins. The bacterial endotoxin specification of a product can be calculated using the dose given. The dose of vaccine people receive is very small. The internationally accepted pyrogenic threshold11 for injectable medicines is 5 EU per kg of body weight, per hour.
The endotoxin limit for a product is usually calculated based on a 70 kg person. At 5 endotoxin units per kg, this means the patient could safely receive up to 350 EU. The value obtained in our testing of every mRNA vaccine batch to date was less than 5 EU/mL. The dose given is also less than 1 mL.
Using the same scenario, a 5 kg child would be allowed up to 25 EU, and lower doses are given to children. Given a 0.25 mL dose of a batch tested at TGA to < 5 EU/mL, this child would only receive up to 1.25 EU, which is 20 times less than the internationally accepted limit of 5 EU/kg/h.
The results are reported as < 5.0 EU/mL (less than 5.0), because the values obtained are below the level of detection of the assay for the dilution used (Table 3 and Table 4).
| AUST R | Number of Batches* | Results | |
|---|---|---|---|
| SPIKEVAX | 370599 | 44 | < 5.0 |
| 388244 | 2 | < 5.0 | |
| 389513 | 7 | < 5.0 | |
| 399553 | 10 | < 5.0 | |
| 418911 | 6 | < 5.0 |
| AUST R | Number of Batches* | Results | |
|---|---|---|---|
| COMIRNATY | 346290 | 80 | < 5.0 |
| 377110 | 2 | < 5.0 | |
| 377111 | 15 | < 5.0 | |
| 393433 | 2 | < 5.0 | |
| 394890 | 5 | < 5.0 | |
| 400874 | 10 | < 5.0 | |
| 419330 | 5 | < 5.0 | |
| 419332 | 1 | < 5.0 | |
| 419371 | 1 | < 5.0 | |
* Batch numbers tested may not reflect total number of batches released in Australia. Some batches:
| |||
Conclusion
All batches of mRNA vaccines tested for residual DNA content in the TGA Laboratories were found to comply with the WHO recommended limit for residual DNA of up to 10 ng per dose, consistent with the results reported by manufacturers.
Current mRNA manufacturing processes, as evaluated and approved by the TGA, include breakdown of plasmid DNA and additional purification strategies. These processes are effective in controlling levels of residual plasmid DNA to ensure compliance with residual DNA guidelines.
To date, all batches of Comirnaty and Spikevax released to the Australian market have been tested for bacterial endotoxin content by the TGA Laboratories. All batches were found to comply with the registered bacterial endotoxin limit for the particular product. This is consistent with the results reported by manufacturers as reviewed as part of the batch release process. The assay tests these products to the level of 5 EU/mL and every batch demonstrated levels below this limit.
Footnotes
- Guidelines for independent lot release of vaccines by regulatory authorities.doc
- The TGA Laboratories first gained independent laboratory accreditation for testing in the 1980s. The current international standard is AS ISO/IEC 17025:2018 General requirements for the competence of testing and calibration laboratories
- WHO Informal consultation on the application of molecular methods to assure the quality, safety and efficacy of vaccines
- Yang, H (2013) Establishing Acceptable Limits of Residual DNA, PDA J Pharm Sci and Tech, 67 155-163
- Paul Ehrlich Institut, 2023: Methodology of testing COVID-19 mRNA vaccines for alleged contaminations
- Pharmacopoeias | Therapeutic Goods Administration (TGA)
- Spiking is when a known amount of the substance of interest is added to the sample - ICH Q2(R2) Validation of analytical procedures - Scientific guideline | European Medicines Agency (EMA)
- The method can be found at https://www.tga.gov.au/sites/default/files/2024-06/FOI%204878%20Documents_0.pdf page 90
- A 95% confidence interval (CI) for expanded uncertainty is a statistical estimate of the range likely to contain the true value of a measured parameter, and accounts for inherent uncertainty present in the measurement process.
- AUST R is the registration number on the Australian Register of Therapeutic Goods.
- The pyrogenic threshold is the level of endotoxin activity at which a substance is considered to induce fever, typically measured as 5 endotoxin units (EU) per kilogram of body weight per hour for intravenous or intramuscular administration (Calculating Endotoxin Limits for Drug Products | American Pharmaceutical Review - The Review of American Pharmaceutical Business & Technology).
Page history
- Updated two figures in Table 4 for Number of Batches for AUSTR 400874 and 419330.
- V1.1: Minor edits to the Executive summary for readability and clarity.
- Author: Laboratories Branch
- Effective date: November 2024
- V1.0: Original publication
- Author: Laboratories Branch
- Effective date: November 2024
- Updated two figures in Table 4 for Number of Batches for AUSTR 400874 and 419330.
- V1.1: Minor edits to the Executive summary for readability and clarity.
- Author: Laboratories Branch
- Effective date: November 2024
- V1.0: Original publication
- Author: Laboratories Branch
- Effective date: November 2024