Substituting equivalent herbal extract ingredients in listed medicines
This guidance explains when a herbal extract can be substituted as an 'equivalent' ingredient in a therapeutic good, without being considered a separate and distinct good.
Purpose
This guidance document describes the circumstances under which a herbal extract may be considered to be 'equivalent' to an ingredient currently included in a therapeutic good, and which therefore may be used as a substitute for the ingredient, without the product being considered to be a separate and distinct good.
This document also outlines information that, although not currently required to be recorded in product applications, is desirable for sponsors / manufacturers to collect, record, and consider when formulating products, and when determining whether separately sourced ingredients are interchangeable.
Introduction
Due to fluctuations in the availability of extracts, some sponsors / manufacturers at times choose to accept extracts with different extraction profiles, and interchange these with nominated ingredients in product formulations.
Given that different solvents, solvent concentrations and extraction methodology may result in ingredient preparations with different safety and efficacy profiles, the question arises as to what variation, if any, should be permitted for extracts of the same herbal substance.
Factors that impact upon equivalence of extracts
It is recognised that there are a number of factors that can ultimately affect equivalence of extracts, including natural variations in starting material, solvent type and concentration, temperature, pressure, and time.
Table 1 outlines the influence of manufacturing and quality parameters on the quantity of extract and the spectrum of components.
| Parameter | Quantity of extract (native extract ratio) | Spectrum of components |
|---|---|---|
1. Herbal material type plant part water content grade of comminution |
+ + + # |
+ + |
2. Extraction solvent type concentration quantity flow rate* |
+ + + (maceration) |
+ + + (maceration) |
3. Manufacturing procedure/extraction type (maceration/percolation) duration temperature pressure |
+ # + + |
+ # + + |
4. Equipment filling height statical pressure* batch size (extractor, evaporator, dryer) |
+ |
Key
+ This parameter impacts on the quantity of extract or the spectrum of components
# no influence provided that steady-state or exhaustive extraction is reached
* flow rate influences speed of extraction when percolation as a method of manufacture is used
# no influence provided that steady-state or exhaustive extraction is reached
* statical pressure influences speed of extraction
Factors affecting the spectrum of components extracted from herbs
It is particularly important to consider those factors influencing the spectrum of components obtained from the herbal material, as these may affect the quality, safety and efficacy of the final extract.
Where the component profile of an extract is significantly different, extracts should not be interchangeable.
Herbal material
Primary factors that affect the spectrum of components extracted from a given herbal material, are type (botanical species), and plant part. This information is currently recorded in product applications, and if either parameter is changed, the result is a separate and distinct good.
Extraction solvent
The type, concentration and quantity of extraction solvent, will all affect the spectrum of components obtained from a given amount of herbal material. Currently, sponsors are required to nominate both type and concentration of solvent in product applications, but not the quantity of solvent used.
A change to the type of solvent used to make an extract is currently deemed to result in a separate and distinct good. However a limited degree of variation in solvent concentration is now considered to be acceptable, and is unlikely to affect the spectrum of components extracted. (See Permitted variation in factors impacting on quantity of extract below.)
The TGA does not collect data (in product applications) in relation to the quantity of solvent used to make an extract. This factor will require special consideration when determining whether a replacement extract is equivalent to the extract currently included in a therapeutic product.
Manufacturing procedure
The type of extraction procedure used to manufacture an extract, including batch size, conditions of temperature, and pressure, have all been identified as factors that influence the spectrum of components obtained.
The TGA does not collect any data for herbal ingredients in relation to extraction type, temperature or pressure. These factors will require special consideration when determining whether a replacement extract is equivalent to the extract currently included in a therapeutic product.
Permitted variation in factors affecting quantity of extract
A number of factors have been identified as influencing the quantity and composition of the native extract obtained. Differences between extracts may, in part, be reflected in the expression of the native extract ratio.
However, factors affecting the quantity of the native extract obtained, may not necessarily affect the spectrum of components extracted. Therefore it is possible to have a change in the quantity of the extract obtained from a particular batch (reflected as a change in the native extract ratio), which may have no (or little) impact on the qualitative and quantitative composition of the components in the final extract.
Herbal material
Due to the natural variation in the composition of a herbal starting material (raw herb), the native extract ratio may vary from batch to batch. That is, herbs sourced at different times of the year, or from different climactic / geographical situations, may provide differing amounts of extractable herbal components (using defined extraction solvents, and a validated extraction procedure). Over a period of time, sufficient batch values may be obtained to provide an expected range in the native extract ratio, although this may take several harvests, and years of production, to determine.
Any variation in native extract ratio, therefore, may only be determined retrospectively.
Extract solvent
Where the type and amount of solvent used to manufacture a particular extract is the same, a limited degree of variation in solvent concentration is now considered to be acceptable. The ranges outlined in Table 2 below, are considered unlikely to affect the spectrum of components extracted.
The permitted range in solvent concentration uses the minor solvent as a reference point. Although the range is +/- 50 per cent of the minor solvent for solvent concentrations of 1-20 per cent, the acceptable solventvariation for the range between 30-50 per cent is restricted to no more than 10 per cent variation around the minor solvent (as this is the range that could result in significant variation and have implications for phyto-equivalence).
| Minor solvent concentration | Acceptable solvent range |
|---|---|
| 1% | 0.5 - 1.5% |
| 5% | 2.5 - 7.5% |
| 7.5% | 3.75 - 11.25% |
| 10% | 5 - 15% |
| 15% | 7.5 - 22.5% |
| 20% | 10 - 30% |
| 30% | 20 - 40% |
| 40% | 30 - 50% |
| 50% | 40 - 60% |
Manufacturing process
All parameters of the manufacturing process can affect not only the quantity of extract obtained, but also the composition. It is the responsibility of the sponsor to ensure that any variation does not result in a significantly different extract, and hence a separate and distinct good.
Extract ratio
Variation in native extract ratio can result in a variable amount of herbal material used in an extract. In some instances, variation in the equivalent dry weight of a herb used in herbal preparation in a medicine can affect the validity of the amount of ingredient declared on the label, as well as impacting on the recommended dose of the product.
Where a large proportion of extractable material is obtained from a herbal material, the native extract ratio will be low. For example, a low native extract ratio of 2:1 indicates that 50 per cent of the extractable matter obtained from the herb is represented in the final extract.
e.g. 2 kilogram (kg) of herb will provide 1 kg of native extract.
However, when only a small amount of extractable material is obtained using a particular extraction profile, the native extract ratio will be high (e.g. a native extract ratio of 20:1 indicates that only 5 per cent of extractable components are obtained).
e.g. 20 kg of herb is required to obtain 1 kg of native extract.
Whilst a high native extraction ratio is generally reflective of a targeted extraction procedure (i.e. specific components or component classes are selected for), there are instances where a high extraction ratio may simply reflect a partial extraction procedure.
Consideration should be given to ensuring that these extracts are not marketed in a manner that implies that they are 'better' because they are derived from a larger quantity of raw herbal material.
There are also situations where an extract with a high native extract ratio is diluted with a carrier or diluent, for a variety of purposes. The addition of diluents and carriers should always be taken into account when considering whether two extracts are equivalent.
'Not significantly different' / 'Essentially the same'
In order to determine whether or not a herbal preparation is 'not significantly different', or 'essentially the same', it is first necessary to establish a base value from which variance may be considered. When comparing two extracts to determine if they are 'not significantly different / essentially the same', it is important to take account of the extent of dilution of the native extract with carrier(s). If it is not possible to establish equivalence of an extract or preparation via process controls (as described in the sections above), qualitative and quantitative comparison of the original extract, and the intended replacement, may be made using profile chromatograms of the native extract.
Comparison of two extracts for the purposes of determining the degree of similarity or difference must include a quantitative assessment as well as a qualitative assessment of the chromatographic profiles.
Profile Chromatograms2 (developed from Section 4.7 ARGCM Part III)
A profile chromatogram or, as it is more commonly known, a 'fingerprint' chromatogram, is a chromatographic profile of a botanical raw material, a preparation of a herbal material, or other substance that can be compared with that of a reference sample or standard.
Where a profile chromatogram is used to determine whether a particular herbal preparation is'not significantly different' / 'essentially the same' with another herbal preparation, the profile must be unique to the substance, and comprehensive enough to provide a basis for assuring the identity and consistency of the substance with which it is being compared. When determining whether or not two extracts or preparations are 'essentially the same', profile chromatograms should be performed on the native extract, without the addition of excipients.
The profile chromatogram should initially aim to reflect the possible variation that may occur for a substance, so sponsors should ensure that profile chromatograms represent, as far as practicable, as wide a range as possible of a substance's variability. This may involve an investigation of the profiles of substances from different sources, and for botanicals, possibly different seasons. This is most relevant if there are concerns that the safety or quality could be compromised by the source of the substance. Where the literature indicates that potential substitution or adulteration is possible, then the conditions and techniques used to develop the profile chromatogram should also enable the detection of adulterants and differentiation from substitutes.
In developing a profile chromatogram for use when determining whether two herbal preparations are 'essentially the same', sponsors need to give consideration to substances that will not be determined as part of the profile. The profile chromatogram should include all relevant component groups, not just those that are thought to be responsible for the activity of the ingredient (e.g. starches or sugars). If known, and where practicable, a profile chromatogram should therefore be accompanied by information about any constituents in the substance that are not profiled.
Justification for not profiling these other constituents should be provided on the basis that these other constituents have no effect on the identification of the substance.
Profile development
No single technique can be recommended for developing a profile chromatogram. Sponsors should first undertake a rigorous literature search to ensure that profile chromatogram conditions have not already been developed by other researchers. Sponsors can assess the most appropriate technique to use by considering the nature of the major or significant constituents of the substance; for example, volatile oils in a substance would be better determined by gas chromatography (GC) than high pressure liquid chromatography (HPLC), whereas thin layer chromatography (TLC) may be more appropriate than HPLC for determining sugars in a substance.
In developing a profile chromatogram, sponsors may need to experiment with the different chromatographic techniques by using different solvents (including extraction solvents) or elution conditions, different stationary phases and different detection or derivatisation techniques. The techniques and conditions used to develop a profile chromatogram should be optimised to produce the maximum amount of information. In addition, sponsors may wish to combine techniques to obtain more detailed profiles of a substance. In general, techniques and procedures should be:
- objective and reproducible
- tailored to suit the characteristics of the components that are the target of the determinations
- elective enough to separate the components that, as far as is known, are characteristic of the substance
- sufficiently general to profile as many components as possible (more information is better than less)
- robust enough to ensure that labile or unstable components are identified, particularly where a substance's stability is concerned
- optimised to produce high-quality profile chromatograms (texts are available that provide assistance on optimising chromatographic separations).
Sponsors should be aware that a representative profile chromatogram and the techniques and conditions for developing these chromatograms would be publicly available. This is to ensure that substances used in complementary medicines are of suitable quality.
Profile Chromatogram Interpretation
The interpretation of profile chromatograms involves:
- developing profile chromatogram specifications from chromatograms of material of acceptable quality
- comparing and contrasting the size, shape and distribution of relevant peaks or spots in sample and in standard or reference chromatograms
- assessing these differences and similarities against the profile chromatogram specifications to determine compliance with the specification.
Before any sample of a substance can be assessed against standard material, the specifications with which future samples will need to comply must be determined. For profile chromatograms, this approach involves determining the key or indicative peaks/spots and then developing tolerances that can then be used for assessing samples of the substance. Please note that this process may need to be undertaken at several different wavelengths to ensure that all relevant components/component groups that may be used to establish preparation equivalence, have been identified.
To develop these tolerances it may be necessary to examine profile chromatograms of:
- degraded or poor quality material containing the substance, as this will provide an indication of the peak or spot changes associated with a sub-standard substance
- the substance 'spiked' with known adulterants or substitutes, as this will provide an indication of the specificity of the method.
The key or indicative peaks / spots are those that are associated with the degradation of the substance and / or the presence of adulterants or substitutes. In selecting these, sponsors should not focus unduly on one type of constituent (e.g. flavonoids). Sponsors should work from the general to the specific, rather than focusing on specific constituents at the outset.
The size, shape and distribution of the responses can be used to determine profile chromatogram specifications. Sponsors may also wish to consider the ratios of certain responses and not just the individual responses for constituents. Ratios can sometimes represent better indicators of quality because they allow controls to be determined for more than one constituent and may be particularly useful where more than one substance is therapeutically active.
The allowable extent of variations in profile chromatograms will need to be determined on a case-by-case basis. This is because slight variations can be of importance, particularly if the variation is associated with the presence of one or more toxic substances. Conversely, gross changes may sometimes be of limited significance.
As a starting point, sponsors should consider specifications that limit:
- any changes in component responses that are greater than +/- 10 per cent, where the responses are associated with constituents of known therapeutic activity
- any changes in component responses that are greater than +/- 20 per cent, where the responses are associated with constituents that are unknown or are not linked to therapeutic activity.
Sponsors can adopt wider specification limits where these can be justified. Large variations in profile chromatogram specifications should not be used as a means of legitimising substandard material. Specifications should be sufficiently broad to allow for normal variations in the constituents of the substance.
The analyst should note any similarities and differences between the chromatograms obtained from the sample and the reference sample, particularly for any components identified in the specifications. Similarities areas important as differences and should be recorded, particularly where the sponsor is aware that a peak or spot is associated with a constituent of therapeutic or toxicological significance. Differences in responses that exceed the +/- 10 per cent or +/- 20 per cent criteria discussed earlier should be explored and the acceptance of such samples in a finished product must be justified.
Summary
Factors such as the natural variability of the herbal material (in particular the total extractable matter), combined with the solvent system, extraction method and extraction conditions, can all have a significant impact on the quantity and composition of a herbal extract. A change in any one of these factors is generally reflected in the native extract ratio.
In a production environment the native extract ratio will always vary to some degree on a batch to batch basis due to the natural variability of the herbal material. In reality, the acceptable range for the native extract ratio can only be determined retrospectively, on a case by case basis, by controlling all intrinsic factors such as solvent system, extraction method and extraction conditions, and by subjecting a number of production batches to statistical analysis.
A minor variation in solvent concentration ratio is permitted, however as most factors that affect the quantity of extract obtained will also affect the composition of the extract, it is the responsibility of the sponsor and the manufacturer to ensure that any variation in extract parameters does not result in a significantly different extract.
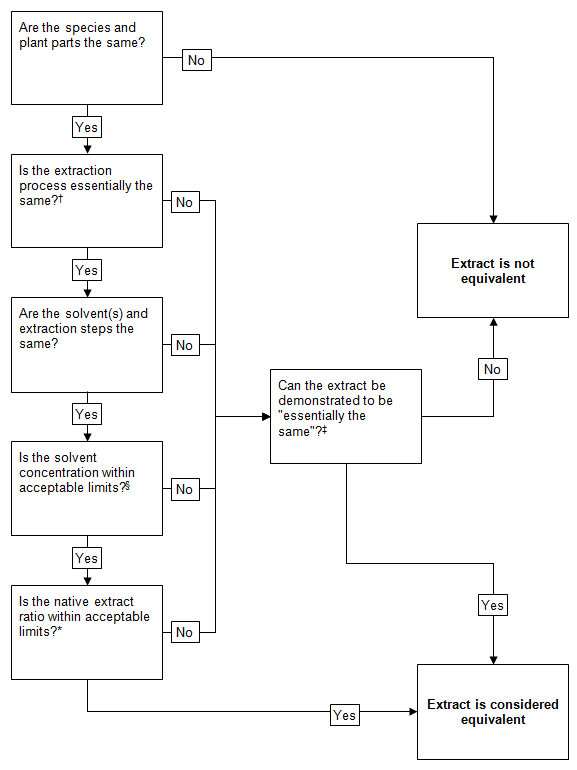
Flowchart: Equivalence of extracts
{kind=link}
This image shows a flowchart for determining if an extract is considered equivalent. The flowchart consists of a series of yes/no decision points, represented by rectangular boxes with questions inside, connected by lines and arrows indicating the flow of decision-making.
The flowchart begins with the question "Are the species and plant parts the same?" If the answer is no, it leads directly to "Extract is not equivalent." If yes, it proceeds to the next question.
Subsequent questions include:
- "Is the extraction process essentially the same?"
- "Are the solvent(s) and extraction steps the same?"
- "Is the solvent concentration within acceptable limits?"
- "Is the native extract ratio within acceptable limits?"
Each "No" answer at these stages leads to "Can the extract be demonstrated to be 'essentially the same'?" If this cannot be demonstrated, the extract is considered not equivalent.
If all questions are answered "Yes," or if the extract can be demonstrated to be essentially the same despite some differences, the flowchart concludes with "Extract is considered equivalent."
§ Some variation is permitted in these parameters. Refer to Table 2.
* Some minor variation may be permitted following retrospective consideration over a number of batches.
‡ Refer to guidance on 'not significantly different/essentially the same' in the body of this document.
† See Table 1.
Glossary
The following definitions apply to this guideline:
| Term | Definition |
|---|---|
| Excipient | Means an ingredient other than the active ingredient. Excipients may be added to extracts in order to adjust the concentration; enhance stability; limit microbial growth; and to improve drying, flow, or other manufacturing characteristics. |
| Extract | Means the complex, multi-component mixture obtained after using a solvent to select for, or remove, components of the herbal material. Extracts may be in dry, liquid or semisolid form. Extracts are not the same as expressed juices, pure chemicals isolated from a herb or synthetically modified plant constituents. |
| Herbal material | Means a plant or part of a plant (defined by its botanical scientific name according to the binomial nomenclature system and by the plant part), whether whole, fragmented, cut or ground, and in an unprocessed state (whether fresh or dried). |
| Herbal preparation | Means any preparation of a herbal material that involves any further processing of the raw herb other than drying, fragmenting, cutting or grinding. |
| Native extract | Means the material consisting only of components present in the original plant or formed during the extraction process, excluding any excipients or other added substances. This term may refer to liquid extracts or semi-solid extracts from which the added solvent has been removed, or may refer to a dry extract or that portion of a finished extract that is comprised solely of plant components. |
| Native extract ratio | Means the ratio of the mass of herbal material to the mass of the resulting native herbal preparation (= native extract). In summary, 'native' extracts consist solely of extractable herbal matter, whilst 'extracts' or 'herbal extracts' contain substances other than herbal extractable matter, excipients needed for adjustment purposes (such as standardisation), or residual extraction solvents. It is extremely useful to be able to differentiate between an 'extract' and a 'native extract', both in terms of avoiding some of the misunderstandings that currently exist regarding the quantification of extracts on product specifications, certificates of analysis, and on labels, as well as in terms of allowing an unambiguous description of the 'ingredient' for quality purposes. When determining whether two similar extracts are in fact equivalent, the native extracts should be used for the comparison. |
Footnotes
- This table was based on information included in Gaedcke F., Steinhoff B, & Blasius, H. Herbal Medicinal Products: Scientific and Regulatory Basis for Development, Quality Assurance and Marketing Authorisation, Medpharm Scientific Publishers, Stuttgart 2003.
- This section on profile chromatograms should be considered in conjunction with the information set out in Part III, Section 4.7 of the Australian Regulatory Guidelines for Complementary Medicines (ARGCM).
Page history
Title changed from 'Guidance on equivalence of herbal extracts in complementary medicines' to 'Substituting equivalent herbal extract ingredients in listed medicines' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Original publication.
Title changed from 'Guidance on equivalence of herbal extracts in complementary medicines' to 'Substituting equivalent herbal extract ingredients in listed medicines' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Original publication.