IVD sponsors: a roadmap to market
This road map presents an overview of the requirements for supplying an IVD for Australian sponsors.
This road map is for Australian sponsors of In Vitro Diagnostic (IVD) medical devices.
You will find out the requirements for supplying and IVD under the medical devices regulatory framework.
The appropriate pathway is determined by the manufacturer's classification of the IVD.
IVD overall process diagram
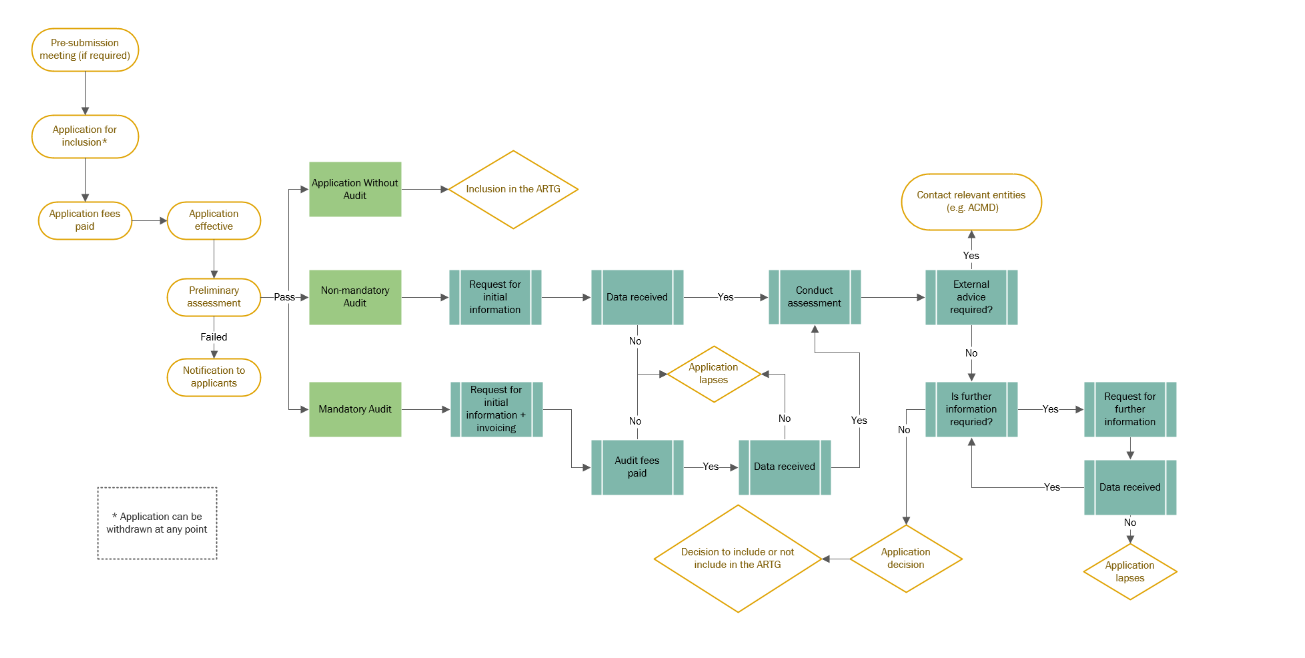
{kind=link}
Footnotes (and links to further information)
How we regulate IVDs
An IVD is any medical device which is a reagent, calibrator, control material, kit, specimen receptacle, software, instrument, apparatus, equipment or system, whether used alone or in combination (with other diagnostic goods for in vitro use), intended by the manufacturer to be used in vitro for the examination of specimens derived from the human body, solely or principally for the purpose of giving information about a physiological or pathological state, a congenital abnormality or to determine safety and compatibility with a potential recipient or to monitor therapeutic measures.
Sponsors applying for an eBusiness Services account
Applications for inclusion on the Australian Register of Therapeutic Goods (ARTG) are submitted electronically through the TGA Business Services (TBS) portal. New sponsors will need to establish an eBusiness Services account to access eBusiness Services.
Sponsor obtains the IVD classification from the manufacturer
The manufacturers must classify the IVD according to the classification rules, based on the intended purpose of the IVD.
Sponsor obtains the GMDN from the manufacturer
The Global Medical Device Nomenclature (GMDN) is a collection of internationally recognised terms, each with a unique number that is used to accurately describe and catalogue medical devices. It is the responsibility of the manufacturer to assign the appropriate GMDN.
- The use of GMDN codes for IVD medical devices in Australia
- For background information about GMDN codes refer to the GMDN agency website - external site.
Requirements for manufacturer's evidence
Australian manufacturers of IVD medical devices are required to have acceptable conformity assessment evidence based on the classification of the device. The evidence for class 2 to 4 IVDs can include conformity assessment certificates issues by the TGA or evidence from a comparable overseas regulator.
- Manufacturer evidence for IVD medical devices
- TGA Conformity Assessment overview
- Comparable overseas regulators of medical device applications
Declaration of Conformity
It is strongly recommended that sponsors obtain the manufacturer’s Declaration of Conformity before applying to include a medical device on the ARTG.
- Details of what should be included in the declaration can be found in the Australian Declaration of conformity templates
- Essential Principles
- TGA Conformity Assessment overview
Conformity Assessment procedure for IVDs
Manufacturers of IVD medical devices may choose to seek conformity assessment certification from the TGA.
Sponsors submits manufacturer's evidence through eBusiness Services
Only the QMS certificate should be submitted as manufacturer’s evidence. Any other information such as the Declaration of Conformity and other information will be requested by the TGA when required. When submitting new evidence through eBusiness Services, ensure ALL product GMDNs applicable to the scope of that evidence are entered prior to finalising the submission. Once manufacturer’s evidence has been accepted, details relating to the manufacturer, sponsor and GMDN codes will be linked to the medical device application as it is submitted through eBusiness Services.
IVDs of the "same kind of medical device" can be included by a sponsor under a single medical device application
The "same kind" of medical device has:
- same sponsor
- same manufacturer
- same GMDN
- same risk classification
Class 4 IVDs are only considered to be of the "same kind" if in addition to the above, they also have the same Unique Product Identifier (UPI)
Note: Companion Diagnostics that are Class 3 IVD medical devices also have the same UPI.
Sponsor submits an IVD application through eBusiness Services (application fee applies)
When lodging an application, the sponsor must certify the following:
- The product is a medical device and is intended for a specific purpose
- The IVD is correctly classified according to the rules
- The IVD complies with the Australian Essential Principles for quality, safety and performance, and information is available to substantiate compliance
- An appropriate conformity assessment procedure has been applied to the IVD, and sufficient information is available to substantiate the application of the conformity assessment procedures
- The IVD complies with advertising requirements
- The IVD does not contain any substances prohibited from import into Australia
- All information included in or with the application is complete and correct
- These are procedures in place with the manufacturer to obtain and submit to the TGA any information required, within period specified in the request for information. This may include evidence of compliance, the declaration of conformity, etc. (refer to the Therapeutic Goods Act 1989, Section 41FD)
Also see:
Application audit of IVD medical devices
As per our regulation, the following high-risk IVD medical devices will undergo mandatory application audit, if they are NOT supported by comparable overseas regulator certification:
- IVDs used for self-testing
- Point of care tests
- Class 3 IVDs
- Class 4 IVDs
- Class 4 in-house IVDs
Any other application may be selected for a non-mandatory application audit.
- Comparable overseas regulators for medical device applications
- Application audit (Technical file review) of IVD medical devices
Sponsors are only invoiced an assessment fee for IVDs undergoing a mandatory application audit
Applications that are selected by the TGA for a non-mandatory application audit do not attract an assessment fee.
TGA includes the IVD on the ARTG
A public summary of the IVD will be available on the Australian Register of Therapeutic Goods (ARTG).