Understanding the application requirements for a new substance in listed medicines
Guidance on the application process and information required for a substance to be evaluated for use in listed medicines.
Purpose
The Application requirements for new substances in listed medicines (ARNS), provides guidance for applicants requesting evaluation of a substance for use in listed medicines [AUST L listed medicines and AUST L(A) assessed listed medicines]. It replaces guidance formerly included in the ARGCM V 8.0 Part C.
The guidance includes:
- Application process for new substances
- How the TGA regulates market exclusivity of a new substance before and after approval
- Administrative, quality and safety information requirements for a substance application
- A summary of core information requirements for different substance types
Legislation
Abbreviations
Refer to the TGA acronyms and glossary for terms, definitions and acronyms.
Section A – Application process for new substances for use as ingredients in listed medicines
Information: This guidance replaces archived ARGCM V8.0 Part C: Evaluation of a substance for use in listed complementary medicines. Details of the Information required in an evaluation of a substance for use in listed medicines is now in Section B – Information requirements.
Refer to Standards, guidelines and publications for a list of all guidance relevant to listed medicines and registered complementary medicines.
Background information
Applications to vary the Permissible ingredients determination
All listed medicines (AUST L listed medicines, and AUST L(A) assessed listed medicines) may only contain ingredients included in the Therapeutic Goods (Permissible Ingredients) Determination (the Permissible Ingredients Determination). The Permissible Ingredients Determination is a legislative instrument made by the Minister for Health under section 26BB of the Therapeutic Goods Act 1989 (the Act).
For a new substance to be included or an existing substance to be varied in the Permissible Ingredients Determination, an applicant must make an application to the Secretary under section 26BD of the Act for a recommendation that the Minister vary the Permissible Ingredients Determination. Applicants must use the application form to set out the recommendation sought by completing the approved application form in TGA Business Services. An application can be submitted for:
- A new medicine substance not currently included in the Permissible Ingredients Determination
- A proposed new role or a change to the existing requirements for use of a current permitted ingredient, for example:
- for an ingredient permitted for use as an excipient to be used as an active ingredient
- to change the permitted level of use
- to change the permitted route(s) of administration
The TGA will evaluate the substance taking into account whether it is of appropriate quality and safety to be permitted for use in listed medicines. A TGA delegate of the Secretary of the Department of Health and Aged Care must either, make a recommendation (successful applications) or refuse to make the recommendation (rejected applications) to the Delegate of the Minister to vary the Permissible Ingredients Determination.
If the substance is determined to be sufficiently safe and of appropriate quality, it will be recommended for inclusion in the Permissible Ingredients Determination and the Delegate of the Minister may vary the instrument. In some cases, ‘requirements’ (for example, restrictions on the concentration or maximum daily dose) may be attached to the use of the ingredient in a listed medicine so that safety and quality can be maintained.
Once an ingredient is included in the Permissible Ingredients Determination, it may be used in any listed medicine provided any requirements for use are complied with.
Substances eligible for evaluation for use in listed medicines
A substance may be eligible for evaluation for suitability for use as an ingredient in listed medicines if:
- the substance is not a prohibited import
- the substance or its component(s) (e.g. components of herbal substances) is/are not included in a Schedule to the Poisons Standard
Some substances are subject to the conditions of a Schedule (or applicable Appendix) to the Poisons Standard only if present in a certain quantity in a finished product. Accordingly, appropriate restrictions (for example, dose or route of administration) must be placed on the use of such an ingredient in listed medicines.
If the proposed new substance is not currently in a Schedule to the Poisons Standard but the substance, or its component, has a potential safety concern that may meet the criteria for inclusion in a Schedule, you should seek advice from the TGA prior to submitting an application. If during the course of the evaluation it is identified that the substance meets the criteria for inclusion in a Schedule, the matter may be referred to the relevant scheduling advisory committee. It may be determined that the substance is not suitable for use in listed medicines on the basis of the scheduling decision.
If you consider the scheduling of a substance should be reconsidered, you can submit an Application to amend the Poisons Standard.
Application categories for evaluation of substances
Applications for evaluation of a substance to be used in listed medicines are categorised into four application levels (IN1, IN2, IN3 and IN4). Each application category has defined submission requirements. An IN4 category requires supporting safety and quality information described in Section B – Information requirements for TGA evaluation. Where an applicant can provide an unredacted evaluation report from a comparable overseas body (COB), or where the quality of the substance is based on a monograph contained in a default standard, less supporting information and shorter evaluation timeframes apply to categories IN1, IN2 and IN3.
Requirements for each application category are described in the Mandatory requirements for an effective application to vary the Permissible Ingredients Determination.
Important: COB evaluation reports that are provided for IN1, IN2 and IN3 applications must be from a list of COBs that is determined by the TGA under Regulation 16GJ – see Comparable overseas bodies (COBs) for complementary medicines.
Timeframes and fees for evaluation of substances
Regulation 16GI of the regulations provides for legislated timeframes for evaluation of a substance for use in listed medicines. The timeframes are provided in Table 1.
Table 1: Timeframes for the evaluation of a substance for use in listed medicines
| Application level | Preliminary assessment (days) | Evaluation (days) |
|---|---|---|
| IN1 | 40 | 70 |
| IN2 | 40 | 120 |
| IN3 | 40 | 150 |
| IN4 | 40 | 180 |
Within 40 working days of receiving an application, the TGA delegate of the Secretary will notify the applicant in writing whether the application has passed preliminary assessment and been accepted for evaluation.
The timeframes for the evaluation of a substance for use in listed medicines:
- only commence once an application is accepted for evaluation and the evaluation fee has been paid
- apply to working days only and exclude public holidays and weekends
- exclude the time when the evaluation clock has stopped (for example: the time taken by the applicant to provide responses to formal requests for information; or when the applicant and TGA agree to a mutual stop clock)
If the Secretary does not make a recommendation within the evaluation timeframe, the TGA must refund 25% of the prescribed evaluation fee.
Applying for evaluation of a substance for use in listed medicines
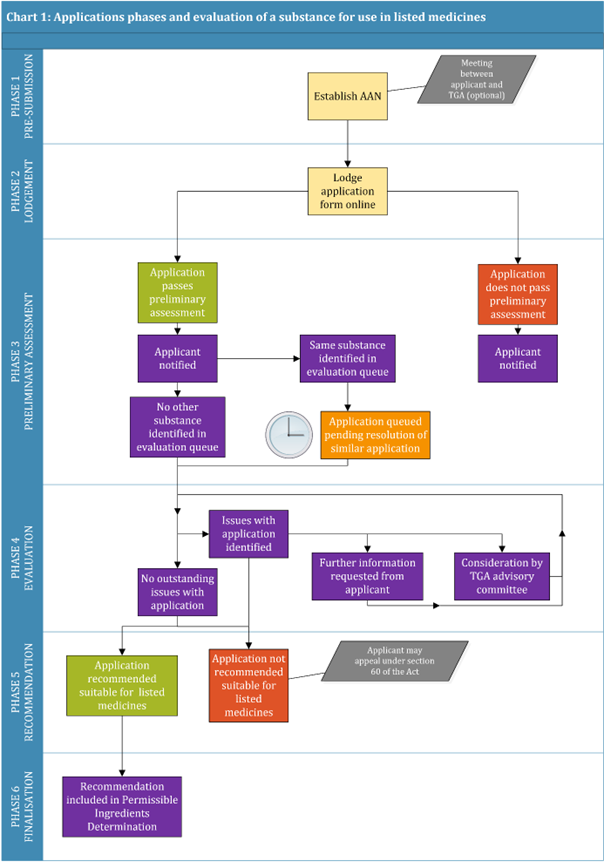
An application for evaluation of a substance for use in listed medicines passes through the following phases:
- Phase 1: Pre-submission phase
- Phase 2: Lodgement of application and payment of application fee
- Phase 3: Preliminary assessment of application
- Phase 4: Evaluation
- Phase 5: Recommendation
- Phase 6: Finalisation
Phase 1: Pre-submission phase
Applying for new Australian approved name
If the proposed new substance does not have an approved name that is selectable from the approved names list using the approved online application form in TGA Business Services, you must submit a proposal for a new name prior to submitting the application for evaluation of the substance - see Application forms for proposing names. You should receive correspondence about the approval status of the proposed substance name. Once this is done, the name will then be selectable in the approved online application form. If the name of the substance is not resolved prior to submitting the application the TGA may be unable to identify if your application is a duplicate of an existing application. See ‘After approval of a new ingredient – exclusive use’ and ‘What if two applicants submit an application for the same ingredient?’ for more information about eligibility for market exclusivity.
Pre-submission meeting
You may wish to request a meeting with the TGA prior to submitting an application for evaluation of a substance for use in listed medicines. See Pre-submission meetings with the TGA for details on arranging a meeting. There is no fee associated with a pre-submission meeting. The TGA may be able to address any questions proposed for the pre-submission meeting in writing in which case a pre-submission meeting will not be required.
The purpose of this is to ensure that you are aware of the legislative requirements and data required for a submission to be accepted for evaluation. If it is determined that the proposed data dossier is likely to not address a mandatory requirement (see Dossier preparation), you have the opportunity to address these deficiencies prior to submitting the application.
Dossier preparation
The information requirements are defined within Mandatory requirements for an effective application to vary the Permissible Ingredients Determination. These information requirements must all be addressed in the dossier to pass preliminary assessment (Phase 3), however some core information categories may be able to be addressed with justifications instead of data.
The content and merit of a justification (i.e. whether lack of data or an alternative approach is appropriate) will be assessed during the evaluation phase (Phase 4).
Section B – information requirements (this document) provides guidance on the types of data that can be provided to meet each core information requirement.
When a justification needs to be provided
If information cannot be provided for a core information requirement defined in Appendix A of Mandatory requirements for an effective application to vary the Permissible Ingredients Determination, or the information does not adhere to a relevant EU or ICH guideline adopted in Australia to establish safety or quality for use in listed medicines, a justification must be provided. The tables in Appendix A state which core information requirements a justification will not be accepted for.
Purpose of justifications
Each justification performs two functions:
- it needs to be present and address why each relevant requirement or guideline is not met or how it is addressed in a different way, in order for the application to be accepted by the TGA for evaluation; and
- once the application has been accepted for evaluation, each justification needs to be sufficient for the TGA to be satisfied that the substance can be included in the Permissible Ingredient Determination.
What needs to be included in a justification
The justification needs to include:
- an explanation of the requirement, guideline or part of the guideline that is not being met, and supporting reasons why it cannot be met with full text citations of relevant papers if applicable; and
- if an alternative approach is proposed, an explanation of the approach and supporting documents where appropriate, to explain why the approach is valid.
Justifications that are not complete and/or sound may result in an application being rejected after evaluation.
Phase 2: Lodgement of application
You are required to submit:
- a completed online application form in TGA Business Services that sets out the recommendation sought for varying the Permissible Ingredients Determination. The information you provide in this form will establish the scope of the evaluation and if successful, will inform the Permissible Ingredients Determination entry for the substance.
- the dossier containing all required technical data for the selected application category according to Appendix A of Mandatory requirements for an effective application for a new substance in listed medicines (further guidance in Section B of this document).
- the application fee (refer to Summary of fees and charges for applicable fees).
Important: All data should be submitted at the time you lodge your application. Omitting relevant data from your application may jeopardise the acceptance of your application or cause unnecessary delays in the evaluation.
The TGA will acknowledge receipt of your application and provide you with an application number that you should reference in all communication on the application.
Phase 3: Preliminary assessment of application
In general, preliminary assessment aims to identify applications that are unacceptable, for example when the dossier is missing core information requirements or justifications as outlined in the Mandatory requirements for an effective application to vary the Permissible Ingredients Determination. Only critical deficiencies in the dossier will be identified at this stage.
During the preliminary assessment, the TGA will also confirm that the correct application level has been selected, all fields on the application form have been completed and the correct application fee has been paid.
Application accepted for evaluation
An application that passes preliminary assessment progresses to the evaluation phase, unless it is a duplicate of another application. See ‘What if two applicants submit an application for the same ingredient?’ for more information.
If your application is progressing to evaluation, you will be notified in writing and an invoice will be issued for the evaluation fee. Your application will lapse if the invoice is not paid within two months. The evaluation process will not commence until the evaluation fee has been paid in full. The evaluation fee will not be refunded if the application is withdrawn during the evaluation phase.
Application not accepted for evaluation
Applications that do not pass preliminary assessment will not progress to the evaluation phase and you will not be invoiced for the evaluation fee. If your application is not accepted for evaluation, you will receive a letter explaining the reasons why. Any other administrative matters in relation to the application will be discussed with you directly. The application fee will not be refunded if your application is not accepted for evaluation.
Phase 4: Evaluation
Safety and quality information will be reviewed to determine if the substance is of sufficiently low risk to be used in listed medicines. The same evaluation process applies for substances proposed for use as active or excipient ingredients, however different quality information requirements for excipients may apply in some circumstances (see Information required to demonstrate quality in Section B).
Important: Efficacy evidence for indications or claims that will be made when a new medicine is listed, is not required as part of a new ingredient application dossier. However, when a permitted ingredient is included in a new medicine, the sponsor of the medicine is required to certify (under section 26A of the Act) that they hold evidence to support the indications and claims made for their medicine. It is a condition of listing that the sponsor of the medicine must provide this evidence to the TGA, if requested to do so. The medicine may be cancelled from the ARTG if any of the sponsor’s certifications under section 26A of the Act are incorrect.
Requests for information
The TGA may make a request for additional information under subsection 26BE(3A) of the Act to clarify or address issues identified during the evaluation. The time between the request being issued and receipt of the applicant’s response will not be counted as part of the evaluation timeframe (the ‘evaluation clock’ will stop). Applicants will be notified of the timeframe for the response.
Evaluators may also seek clarification of minor issues on an informal basis and in these circumstances the evaluation clock will not stop.
The applicant should provide an electronic submission of the requested information. Additional unsolicited data will not be accepted. It is important that applicants respond to a request within the timeframe provided. If the response is not received within the timeframe specified, or if the issues remain unaddressed, the application will proceed to the decision phase without the additional information. This may result in a refusal to recommend the inclusion of the substance in the Permissible Ingredients Determination. Although the TGA may grant extensions to the response due date, this will only be done at the discretion of the delegate if the request is received well before the due date, and if the circumstances are extenuating to justify the need for an extension.
Consideration by a TGA advisory committee
In some circumstances the Minister or Secretary of the Department of Health may seek advice, in relation to the application, from a TGA advisory committee, for example:
- the Advisory Committee on Complementary Medicines
- the Advisory Committee on Medicines Scheduling
- the Advisory Committee on Medicines
You will be informed that a committee’s advice is being sought and given opportunity to provide comment for the committee’s consideration. Any advice by the committee will be included in the evaluation considerations.
Phase 5: Recommendation
After considering the quality and safety of the substance, any response to a request for information from the applicant and any advice from advisory committee(s), the Secretary may:
- make a recommendation that the Minister include the substance in the Permissible Ingredients Determination as requested in the application (successful applications), or
- refuse to make the recommendation sought by the applicant (rejected application)
During evaluation, specific requirements that were not proposed in your application may be determined to be necessary to ensure the safe use of the substance in listed medicines e.g. restrictions on the permitted maximum daily dose, duration of use and vulnerable populations. The exact wording for entry in the Permissible Determination will be determined by the TGA when the evaluation is complete. This is often required to ensure wording within the Determination is consistent and clear. In this instance, the TGA may contact you to confirm your agreement with the outcome and that you wish to amend your application accordingly. If you agree, the Secretary can make a positive recommendation to include the substance in the determination. If you do not agree with the evaluation outcome, the Secretary is likely to refuse to make the recommendation requested. If the application is refused, you will be advised in writing as soon as practicable and provided the reasons why it was not successful.
For information: Applicants requesting an evaluation of a substance for use in listed medicines can appeal the Secretary’s decision to refuse to make a recommendation under section 60 of the Act. Refer to Guidance for requesting reconsideration of an initial decision.
Phase 6: Finalisation
If the Secretary makes the recommendation, a TGA delegate of the Minister will then make a final decision to vary the Permissible Ingredients Determination to include a new ingredient or refuse to do so.
If an ingredient is determined to be suitable for use in listed medicines, the approved ingredient is added to the Permissible Ingredients Determination for use in listed medicines. All permissible ingredients are also made available in TGA Business Services.
If an exclusivity period for use of the ingredient is applicable, this will be included as a specific requirement for the ingredient in the Permissible Ingredients Determination.
Chart 1 illustrates the application stages and simplified evaluation process for the evaluation of a substance for use in listed medicines.
{kind=link}
After approval of a new ingredient - exclusive use
Following an evaluation of a substance, subsection 26BB(2A) of the Act allows the Minister to permit the successful applicant to have exclusive use of that ingredient for a 2 year period (the protected ingredient). The exclusivity period will be specified in the Permissible Ingredients Determination as a ‘requirement’ relating to the use of the ingredient in listed medicines.
What is ‘market exclusivity’?
Market exclusivity prohibits unauthorised sponsors from using an ingredient that has market exclusivity (a protected ingredient) in a medicine listed in the ARTG.
It is intended to reward the resources invested by innovators who research and develop new ingredients to be used in listed medicines. Market exclusivity allows a protected ingredient to only be used in listed medicines by the ingredient owner and any other authorised sponsors.
Other sponsors can use the ingredient to develop a product and prepare for future market authorisation, however they cannot list a product on the ARTG or supply it until the expiry of the 2-year exclusivity period.
Ingredients that are eligible for exclusivity
Exclusivity only applies to new active or excipient ingredients that can be used in listed medicines (including complementary medicines, sunscreens and oral health products).
Exclusivity is only permitted for an ingredient that is not currently included in the Permissible Ingredients Determination, including under a synonym name. That is, the ingredient must be a new item in Table 1 in Part 2 of Schedule 1 to the Permissible Ingredients Determination, provided that it has not previously been evaluated and approved by the TGA for use in listed or registered medicines.
Exclusivity will not apply to applications submitted for a new role or a change to any existing ingredient requirements, for example:
- change from excipient to active use
- update the permitted level of use (e.g. 0.5% to 1%)
- change the route of administration (e.g. from topical to oral use)
- update the plant part, preparation method or purity
- allow for a new strain of an existing species (e.g. LA-5 strain of Lactobacillus acidophilus)
Exclusivity will also not apply to ingredients evaluated as part of a new registered complementary medicine application.
How market exclusivity works
Market exclusivity is optional for successful applicants of newly approved ingredients. It is the responsibility of the applicant to ‘opt in’ at the time of making the application to ensure that the TGA puts the necessary administrative arrangements in place.
During the specified exclusivity period the use of a protected ingredient in a listed medicine will be restricted to:
- the applicant who requested evaluation of the substance (who may or may not be a medicine sponsor)
- other persons authorised by the applicant
The ingredient owner and authorised sponsors will be identified in the Permissible Ingredients Determination by their name and TGA client ID. The exclusivity details will appear as a specific 'requirement' relating to the use of the ingredient in listed medicines (i.e. this will be identified in Column 4 of the Determination). See Diagram C1.
Diagram C1: Example of exclusivity requirement
| Column 1 | Column 2 Ingredient Name | Column 3 Purpose of the ingredient in the medicine | Column 4 Specific requirement(s) applying to the ingredient in column 2 |
|---|---|---|---|
| Example ingredient | A,E | Only to be used in a medicine where [Applicant name] (Client ID 12345), who applied to have the ingredient included has given written authorisation to the sponsor of a medicine to include the ingredient in the medicine. This paragraph ceases to be a requirement for this ingredient after 30 June 2018. |
The exclusivity period starts from the date the ingredient is included in the Determination and ends 2 calendar years later (for example: start 1 July 2018 and end 30 June 2020). At the end of the exclusivity period, the ingredient will become available for any sponsor to include in a listed medicine.
Should an ingredient owner revoke market exclusivity for a third party to use their protected ingredient, then this is a matter to be managed under the agreed commercial arrangements between the two parties. The TGA will not partake in any such proceedings and will not alter any administrative arrangements applied in ELF for the protected ingredient. The TGA will not cancel a medicine, that was legitimately included in the ARTG at the time of listing, based on a subsequent sponsor revocation of market authorisation.
Using exclusive ingredients
Ingredients that are subject to an exclusivity period will be publicly viewable via the Ingredients Table search on the TGA Business Services website and in the Permissible Ingredients Determination. Ingredients will also be viewable in the online application form (Electronic Listing Facility) for listed medicines.
An ingredient subject to an exclusivity period can only be used by the ingredient owner and authorised sponsors to list a medicine in the ARTG.
For information: Ingredient owners can grant access to use an exclusive ingredient to additional sponsors by submitting the ‘Notification of an authorisation to use a protected ingredient’ form when the new substance application is made or after the ingredient is included in the Permissible Ingredients Determination.
New ingredients approved with an exclusivity period are not available for inclusion in Proprietary Ingredients unless authorised by the ingredient owner, but can be used in registered medicines without authorisation.
How does the TGA ensure exclusive use of approved ingredients?
The Electronic Listing Facility contains rules to help ensure that only authorised sponsors can use a protected ingredient.
Use of a protected ingredient within the exclusivity period without an approval from the ingredient owner would contravene the requirement relating to the use of the ingredient and provide grounds to cancel the listing of the medicine from the ARTG under paragraph 30(1)(e) of the Act.
The TGA will not intervene or arbitrate disagreements between sponsors, manufacturers or suppliers in relation to authorisation agreements or competing applications.
What if two applicants submit an application for the same ingredient?
Market exclusivity is awarded to one applicant as the first “innovator”, however it is possible that before a substance is approved, there may be competing applications received by the TGA. To manage competing applications, the TGA will consider valid applications on a first in, first served basis. We may provide informal advice to potential applicants if a substance is already under review during the pre-submission phase. However, ultimately the risk of submitting a competing application will sit with the ingredient applicant.
Regulation 16GI of the Therapeutic Goods Regulations 1990 stipulates how decisions to make a recommendation on an application are considered when there are one or more other applications for the same substance. In the event of a second or subsequent application being received for an eligible substance already under consideration by the TGA, the applicant would be notified during the preliminary assessment phase that there is already an existing application for the same substance. If the second or subsequent application passes preliminary assessment, and the applicant wished to continue with their application, it would be placed in a queue without progressing to evaluation.
If the first application was refused, and any appeal periods exhausted, then the next application in the queue would be informed, invoiced for the evaluation fee, and enter the evaluation phase once the fee is paid.
If the first application resulted in a successful recommendation to include the substance on the Permissible Ingredients Determination, the TGA will contact applicants in the queue and determine if they wish to continue to evaluation or wish to withdraw their application. To ensure the first applicant receives their two-year market exclusivity in full, successful applications to vary an ingredient that is currently under exclusive use would not be included in the Permissible Ingredients Determination until the exclusivity period had expired.
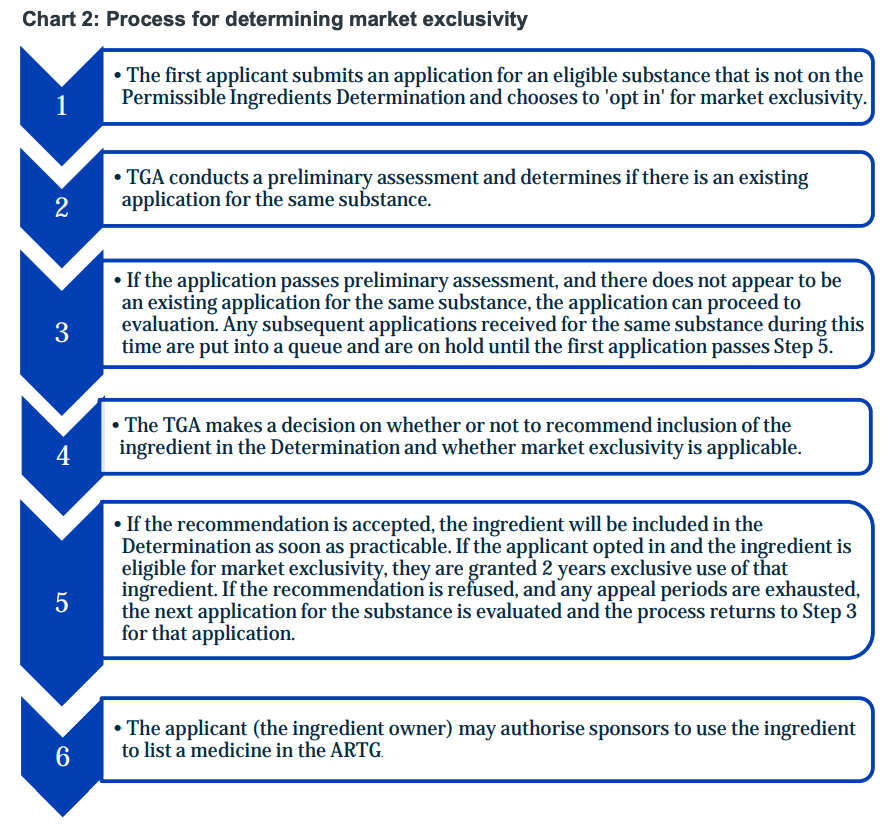
Chart 2: Process for determining market exclusivity
{kind=link}
Process for determining market exclusivity:
- The first applicant submits an application for an eligible substance that is not on the Permissible Ingredients Determination and chooses to 'opt in' for market exclusivity.
- TGA conducts a preliminary assessment and determines if there is an existing application for the same substance.
- If the application passes preliminary assessment, and there does not appear to be an existing application for the same substance, the application can proceed to evaluation. Any subsequent applications received for the same substance during this time are put into a queue and are on hold until the first application passes Step 5.
- The TGA makes a decision on whether or not to recommend inclusion of the ingredient in the Determination and whether market exclusivity is applicable.
- If the recommendation is accepted, the ingredient will be included in the Determination as soon as practicable. If the applicant opted in and the ingredient is eligible for market exclusivity, they are granted 2 years exclusive use of that ingredient. If the recommendation is refused, and any appeal periods are exhausted, the next application for the substance is evaluated and the process returns to Step 3 for that application.
- The applicant (the ingredient owner) may authorise sponsors to use the ingredient to list a medicine in the ARTG.
Section B – Information requirements
Guidance in the section below is intended to complement and provide context to Appendix A of Mandatory requirements for an effective application to vary the Permissible Ingredients Determination.
Administrative requirements
No justifications will be accepted for the absence of a cover letter or table of contents.
Cover letter
Include all the information specified in Table 1 of Appendix A of Mandatory requirements for an effective application to vary the Permissible Ingredients Determination.
The cover letter must also notify us if you are providing a justification for not complying with technical information requirements and/or not adhering to guidelines, and the location of each justification in your dossier. Normally, each justification will be located in the corresponding folder for each core information requirement, and a general overview can be provided in the cover letter.
Table of contents
A comprehensive table of contents is a complete list of all documents in the dossier, and with location references for each document. Provide hyperlinks to each section of data.
The table of contents can be provided for the complete dossier or for administrative, quality and safety data separately.
Each core information requirement in Appendix A of the Mandatory requirements for an effective application to vary the Permissible Ingredients Determination that is applicable to the submission, should have a distinct section in the dossier. For example, applicants may choose to provide one PDF that has separate hyperlinked sections corresponding information requirements; or may choose to provide separate PDF documents or folders that correspond to the information requirements.
Record of any pre-submission meeting or correspondence
Only include information in this section if you had a pre-submission meeting or correspondence with us in relation to your application.
For pre-submission meetings, Include a copy of the pre-submission meeting record.
For pre-submission correspondence via email, include a copy of the relevant email correspondence.
Request for confidentiality
You may request that data contained in your application remain commercially confidential— see Treatment of information provided to the TGA.
Where required, you should identify data that is not in the public domain and may be commercially confidential.
The TGA has an ongoing process to consider and decide whether or not to adopt newly released and updated European Union (EU) and ICH guidelines. As part of this process, some of the above guidelines may be amended, removed or replaced from time to time. Relevant Australia-specific guidelines and adopted EU and ICH guidelines are referenced throughout this document. These effective versions are current at the date this document was published.
Information required to demonstrate QUALITY for substances subject to a monograph in a default standard
For a substance that is subject to a monograph in a default standard [British Pharmacopoeia (BP), European Pharmacopoeia (Ph. Eur.) and the United States Pharmacopoeia – National Formulary (USP)], provide a full un-redacted English copy, or name and version number of the current default monograph in your application.
For a substance that is intended for use as an active ingredient, a batch analysis for minimum, one commercial scale batch (or two pilot scale batches) to demonstrate full compliance with the default standard specifications must be provided. Details of analytical procedures with validation data are not required if all tests are conducted according to the methods in the default standard.
For a substance that is intended for use as an excipient ingredient, a batch analysis is not required. Rather, provide an assurance that the substance meets the full specification in the monograph.
Information required to demonstrate QUALITY for substances not subject to a monograph in a default standard
Quality data for substances not subject to a monograph in a default standard is required to clearly characterise the substance and ensure it is manufactured consistently to a standard that maintains the low-risk status of listed medicines. The amount of information required will vary depending upon whether the substance is classified as a simple or complex substance.
Where a substance is not subject to a specific monograph in a default standard, a list of specifications must be provided in a compositional guideline. A compositional guideline is a list of tests with acceptance criterion and analytical procedure references that is established to ensure quality of the substance for its intended use. It is expected that the manufacturing specifications of the substance will be fully justified with no impurities present at levels exceeding those specified in relevant ICH guidelines. Batch analysis for active ingredients must be provided for minimum two commercial scale batches or three pilot scale batches to demonstrate manufacturing consistency and compliance with the defined specifications in the compositional guideline. Details of analytical procedures with validation data are required for all active substances unless the tests are conducted according to general pharmacopeial methods. Laboratories should hold Good Laboratory Practice (GLP) or accreditation to ISO/IEC 17025 or equivalent, e.g. National Association of Testing Authorities, Australia (NATA), United Kingdom Accreditation Service, American Association for Laboratory Accreditation.
For a summary of core information requirement for different substance types where an ingredient is not subject to a monograph in a default standard, see Appendix (Table 34).
For new drug substances, the following scientific guidelines provide guidance on quality aspects:
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96
- Guideline on the chemistry of active substances CPMP/QWP/130/96 Rev 1
- Guideline on excipients in the dossier for application for marketing authorisation of a medicinal product EMEA/CHMP/QWP/396951/2006
- ICH Q2(R1) Validation of analytical procedures: Text and methodology CPMP/ICH/381/95
Microorganisms
For the purpose of this guidance, microorganisms are defined as whole and intact cells of bacteria and fungi (including yeasts) that are live or non-viable. References to a supplementary guidance document ‘Understanding the application requirements for microorganisms in listed or registered listed complementary medicines’ are made under the relevant sections of this document, and is intended to complement this document. Non-viable microorganisms whose membrane integrity has been compromised such that the cells are no longer intact/whole, are assessed as per other substances. Information required to demonstrate the quality of such substances is outlined in this document.
Dermal excipients
For dermal excipients that are not subject to a monograph in a default standard, a minimum testing specification which includes physical characteristics, identity, assay (unless when only used in a formulation for their physical properties e.g. emulsifier, thickener) and impurity profile must be provided. Batch analysis for minimum, one commercial scale batch or two pilot scale batches must be provided to demonstrate compliance with the specification. A brief description of the manufacturing process is required; however manufacturing process development, process controls, control of intermediates, control of critical steps, and validation of the process are not required for evaluation.
Herbal materials
For herbal materials (unprocessed herbal substance and herbal extracts) that are not subject to a monograph in a default standard, characterisation (including a detailed evaluation of the botanical and phytochemical aspects of the plant, equivalent amount of therapeutically active components (if known and where relevant) or marker compounds and the preparation method used) is essential to develop a compositional guideline that is comprehensive to support the quality and safety evaluation. The quality of a herbal substance is determined by the quality of the raw herbal material, in-process controls, good manufacturing practice controls, and process validation; hence a compositional guideline should specify these critical elements.
The quality of raw herbal materials is determined by such things as:
- botanical characteristics of the plant part
- phytochemical characteristics of the plant part (including biological/geographical variation)
- major components and any significant minor components (analytical/ therapeutic/ toxic)
- identity, assay, limit tests
- cultivation/harvesting/drying conditions (microbial levels, aflatoxins, toxic elements)
- pre/post-harvest chemical treatments (pesticides, fumigants)
- method of preparation, extraction process, including any diluents and extraction solvents
- profile chromatogram and stability of the components
- stress testing to determine potentially harmful degradants
For herbal materials, the following scientific guidelines provide guidance on quality aspects:
- Guidance on equivalence of herbal extracts in complementary medicines
- Quality of herbal medicinal products/ traditional herbal medicinal products EMA/HMPC/201116/2005 Rev. 2
- Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3
- Guidance on the use of modified unprocessed herbal materials in complementary medicines
The general monographs of the British Pharmacopoeia (BP), European Pharmacopoeia (Ph. Eur.) and the United States Pharmacopoeia – National Formulary (USP) are also relevant, for example: the BP monographs ‘Herbal Drugs’, ‘Herbal Drug Preparations’ and ‘Extracts’. Please note that the most recent edition of the cited pharmacopoeia should be used.
Description
Provide a description relevant to the type of substance:
For a chemical substance, provide the molecular formula, molecular structure, Chemical Abstract Service (CAS) registry number (if applicable) and method of manufacture (chemical synthesis and/ or microbial fermentation).
For a herbal material (unprocessed herbal substance and herbal extracts), provide the origin, taxonomy information (including genus, species), plant part, morphology, geographical source, conditions and method of manufacture (cultivated or wild, harvest time, extracted, dried, distilled, purified).
For a substance of animal origin, provide the origin, genus, species, age, animal part, geographical location and method of manufacture (bred or wild, harvest time, extracted, dried, distilled, purified).
For microorganisms, refer to the heading ‘Description’ of ‘Requirements for microorganism characterisation for listed medicines and registered complementary medicines'.
Substances derived from or containing genetically modified organisms
Substances derived from or containing genetically-modified organism(s) (GMO) are regulated under the Commonwealth Gene Technology Act 2000 and Gene Technology Regulations 2001 which includes regulating import, manufacture, transport, storage and disposal. You should contact the Office of the Gene Technology Regulator (OGTR) early in the process of considering importing, manufacturing or supplying a GMO in a therapeutic good. Refer to Types of GMO dealings on the OGTR website or contact OGTR.CDES@health.gov.au. It is the responsibility of applicants to ensure genetically modified substances comply with the provisions of all relevant legislation.
It is necessary to state if the substance is derived from or contains genetically-modified organisms. If the substance is derived from a GMO, or a GMO is used during manufacture, demonstrate (by assay or assessment) absence of this in the final microorganism.
If the substance is a live microorganism that has been genetically-modified, provide a declaration that the organism is exempt under Schedule 2 of the Gene Technology Regulations 2001. Please note that a live GMO that is to be used in a listed medicine does not classify as an 'exempt dealing' because an essential criterion for exemption is that it must be contained. As such, applicants need to seek OGTR approval to be able to import, manufacture or supply the GMO in a therapeutic good. Evidence of OGTR approval is not required in your application however you may provide this information or may be asked to provide this during the evaluation.
Manufacturing details
Description of manufacturing process and process controls
Provide a flow chart of the process that identifies the starting materials, reagents and solvents used, yield ranges and operating conditions for all manufacturing steps.
Provide a sequential, procedural narrative of the manufacturing process, including a detailed description covering the quantities of raw materials, solvents, catalysts and reagents that reflect a representative batch scale for commercial manufacture; critical steps and in-process controls (IPC); equipment; and operating conditions, e.g. temperature, pressure and pH.
Identify any optional/reprocessing steps and provide justification/evidence that they have no significant effect on the final quality of the substance.
Where a manufacturer is unwilling to supply manufacturing details to the applicant, the information can be supplied directly to the TGA with written authorisation from the applicant. In this case, any matters arising from the review of data will be pursued with the manufacturer. The applicant will be notified that matters have been raised with the manufacturer, the details of which will only be provided to the applicant if authorised by the manufacturer.
Herbal materials
The following scientific guidelines provide guidance on quality aspects:
- Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3
- BP General Notices for ‘Herbal Drugs’, ‘Herbal Drug Preparations’ and ‘Herbal Drug Extracts’
For herbal substances, information to adequately describe the plant production and collection, including plant part (e.g. whole, reduced, powdered, fresh, dry), examples of geographical source(s) cultivation, harvesting, drying and storage conditions and batch size, should be provided. Any changes in the manufacturing process, and degradation products produced, may result in a herbal substance that differs from that used to establish safety. The significance of these changes should be considered.
For herbal extracts, extraction steps, extraction ratios and any solvent used, equivalent amounts of therapeutically active components (if known and where relevant) or marker compounds obtained by the extraction process should be provided. Where more than one solvent is used in an extraction step, the concentration of each solvent should be provided. The equivalent dry or fresh weight of the starting material from which the extract was prepared should also be provided.
Chemically derived substances and substances of animal origin
Refer to ‘Guideline on the chemistry of active substances CPMP/QWP/130/96 Rev 1’. Information to adequately describe the manufacturing process, control of materials, critical steps, intermediates, process development and process validation. Particular emphasis should be placed on steps of the process having an impact on the quality of the active substance or intermediates.
Chemically derived excipients intended for dermal use only
Provide a brief description of the manufacturing process. This is expected to provide assurance in terms of the risks that may be associated with the substance and the below sections of manufacturing process development and validation are not required to be evaluated.
Microorganisms
Provide information as per the heading ‘Description of manufacturing process and process controls’ of ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’.
Control of materials
Materials used in the manufacture of the substance (such as raw materials, starting materials, solvents, reagents and catalysts) should be listed identifying where each material is used in the process. Provide the measures used for quality control and release of these materials. These are usually given in the form of specifications or a reference to an acceptable standard, for example: ‘ethanol BP’.
Control of materials does not apply to chemically derived excipients intended for dermal use only.
Controls of critical steps and intermediates
Provide details of critical steps of the manufacturing process and details of how the process is controlled. This information should include tests performed, acceptance criteria and experimental data.
Provide information on the quality and control of any intermediates isolated during the process.
Controls of critical steps and intermediates does not apply to chemically derived excipients intended for dermal use only.
Manufacturing process development
Describe any significant changes made to the manufacturing process of the substance used in producing scale-up, pilot and production-scale batches that may affect the composition of the substance.
Manufacturing process development does not apply to chemically derived excipients intended for dermal use only.
Manufacturing process validation and/or evaluation
Refer to ICH guideline Q11 on development and manufacture of drug substances (chemical entities and biotechnological/ biological entities) EMA/CHMP/ICH/425213/2011 Process validation/evaluation studies should be provided for all types of substances except for chemically derived excipients intended for dermal use only.
Characterisation
The information provided in this section should be used to generate a compositional guideline for the substance that provides a summary of descriptions, tests and appropriate acceptance criteria (which are numerical limits, ranges or other criteria) that define the characteristics and specify the composition of an ingredient permitted for use in listed medicines. When a new ingredient is permitted for use in listed medicines, the compositional guideline for the ingredient is published on the TGA website under current compositional guidelines.
General properties
These are physico-chemical properties (as applicable) relevant to the characterisation of the substance, for example: appearance, colour, state, texture, smell, solubility, particle size, loss on drying, sulphated ash, pH, viscosity, melting point, boiling point, sublimation point, refractive index, microscopic and macroscopic morphology (for herbal materials and microorganisms), residue on ignition etc.
Microorganisms
Provide information as described under the heading ‘General properties’ of ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines'.
Identity
The following scientific guidelines provide guidance on quality aspects:
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96
- Guideline on the chemistry of active substances EMA/454576/2016
- Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3
Identification test(s) confirm the identity of a substance and provides a validated specification that unambiguously distinguish the substance from any other substance or closely related polymorphic forms. More than one test may be appropriate. You must provide:
- a reference to the method corresponding to the identification test(s) employed (e.g. BP Appendix II G. Mass spectrometry (Ph. Eur. 2.2.43), Appendix XI W. HPTLC of herbal drugs and herbal drug preparations (Ph. Eur. 2.8.25); and
- appropriate acceptance criteria for that test (e.g. for HPLC-UV detection, provide wavelength range instead of ‘complies with authenticated reference standard’; for NMR detection, chemical shifts in ppm should conform with the standard/ published reference; for nucleotide sequencing, matches the sequence/ fingerprint for the reference organism).
If the analytical method is not described in a default standard, then validation of that method is required to ensure performance and reliability of that method.
Analytical procedures should be validated in accordance with the scientific guideline, ICH Q2(R1): Note for guidance on validation of analytical procedures: Text and Methodology CPMP/ICH/381/95.
Simple chemical substances
For a simple chemical substance such as a simple salt or single chemical entity, an identification test that is specific for the salt is sufficient. Alternatively, identification test should be specific for individual ions (e.g. bittern compositional guideline).
Complex substances (chemical substances that are ligands and polymorphic forms, herbal extracts and substances of animal origin)
Identification testing should optimally be able to discriminate between compounds of closely related structures (e.g. isomers, enantiomers) which are likely to be present and therefore require specific identity test methods to be considered such as:
- Infrared spectroscopy (IR)
- Gas chromatography (GC)
- Mass spectrometry (MS)
- X-ray powder diffraction
- profile chromatogram (or ‘fingerprint’ that can be compared with an authenticated reference sample or standard), where the separation is based on different principles or a combination of two chromatographic methods into a single procedure, such as HPLC/UV diode array, HPLC/MS, or GC/MS
- Raman spectroscopy
- Optical microscopy
- Nuclear magnetic resonance (NMR)
These tests may include ‘fingerprint’ tests such as TLC, HPLC, IR which must be compared to an authenticated reference standard.
For complex substances, such as herbal preparations, a description of the components with known therapeutic activity, as well as compounds suitable as analytical markers should be provided.
Herbal materials that are not extracts
Herbal materials which are not extracts require identification testing that is specific for the herbal substance and are usually a combination of three or more of the following:
- Microscopic characters
- Macroscopical characters
- Chromatographic procedures (profile chromatogram)
- Chemical reactions
For information on the requirements for identification of herbal substances, refer to Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3 and Identification of herbal materials and extracts.
Information for profile chromatograms
If you are providing profile chromatograms, they should be accompanied by complete details of the extraction steps and procedures (including detectors or detection systems) involved in their production. The information should be of sufficient detail to allow an independent authority to generate the same profile chromatogram.
A profile chromatogram is useful for both qualitative and semi-quantitative assessments. Even in situations where some or all of the components are unknown, profiling can identify variations due to differences in the quality of raw materials, including contamination issues, batch-to-batch consistency concerns and stability issues. If profiling is used semiquantitatively as part of quality control for a substance, for example: it is included in the compositional guideline, consideration would need to be given to the amount of variability that is acceptable.
On its own, a profile chromatogram is not suitable where a component of toxicological or therapeutic activity has been identified in a substance. In this case, specific methods to determine the amount of the toxicologically or therapeutically active component are required.
Importantly, a profile chromatogram may not be indicative of all components within a substance. For example: a profile chromatogram may be generated for the flavonoids in a substance and yet the majority of the substance comprises other components, such as starches or sugars. If known and where practicable, a profile chromatogram should be accompanied by information about the other components in the substance that are not profiled. Justification for not profiling these other components should be provided in the application.
Microorganisms
Provide information as described under the heading ‘Identity’ of ‘Requirements for microorganism characterisation for listed medicines and registered complementary medicines'.
Assay
The following scientific guidelines provide guidance on quality aspects:
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96
- Guideline on the chemistry of active substances CPMP/QWP/130/96 Rev 1
- Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3
Assay tests provide a validated specification to determine the presence and quantity (content) of a specific substance. More than one test may be appropriate. You must provide a reference to the method corresponding to the assay test(s) (e.g. Ph. Eur. method 2.2.29 ‘Appendix III D. Liquid Chromatography’; Ph. Eur. method 2.4.29 ‘Appendix X P. Oils Rich in Omega-3-acids’ for gas chromatography) and the acceptance criteria for that test, which generally includes limits taking into account biological, physical and chemical variation. If the method is not described in a default standard, then validation of that method is required to ensure performance and reliability of that method.
Analytical procedures should be validated in accordance with the scientific guideline, ICH Q2(R1): Note for guidance on validation of analytical procedures: Text and Methodology. CPMP/ICH/381/95.
Chemical substances and substances of animal origin
For chemical substances and substances of animal origin, a specific, stability-indicating method/procedure should be included to determine the content of the substance. In many cases, it is possible to employ the same method/procedure (e.g. HPLC) for both assay of substance component(s) and quantitation of impurities. In cases where use of a non-specific assay is justified, other supporting analytical procedures should be used to achieve overall specificity. For example, where titration is adopted to assay the substance, the combination of the assay and a suitable test for impurities should be used (Refer to section 3.2.1 of ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96).
Herbal materials
For herbal materials (unprocessed herbal substances and extracts) with components of known therapeutic activity or with active markers, assays of their content are required.
Where possible, a specific, stability-indicating procedure should be included to determine the content of the herbal substance. In cases where use of a non-specific assay is justified, other supporting analytical procedures may be used to achieve overall specificity if required.
In the case of herbal materials where the components responsible for the therapeutic activity are unknown, assays of analytical markers or other justified determinations are required. The appropriateness of the choice of markers should be justified. For example, reference to the assay of a marker in the relevant monograph of the European Pharmacopoeia is an appropriate justification (Refer to section 3.2.1 of Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3).
Excipients for dermal use only
Providing an assay is not a requirement when the excipient is only to be used in a formulation for its physical properties (e.g. emulsifier, thickener). Please note that a restriction may be placed so that the excipient will only be limited to use for physical properties in line with the purposes for which the substance was evaluated for.
Microorganisms
Provide information as described under heading ‘Assay’ of ‘Requirements for microorganism characterisation for listed medicines and registered complementary medicines'.
Impurities and incidental constituents
Information in this section should be presented as per the applicable guidelines:
- ICH Q3A(R2) Impurities in new drug substances (CPMP/ICH/2737/99)
- ICH Q3C(R8) Impurities: guideline for residual solvents (EMA/CHMP/ICH/82260/2006)
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances (CPMP/ICH/367/96)
- ICH Q6B Specifications: test procedures and acceptance criteria for biotechnological/biological products (CPMP/ICH/365/96)
- Guideline on Specifications: Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products (EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3)
- Guideline on the chemistry of active substances (CPMP/QWP/130/96 Rev 1)
- SC IV D. Residual solvents, Ph. Eur. general texts 5.4
- Appendix VIII L, Residual solvents, Ph. Eur. method 2.4.24
Impurities and incidental constituents are those constituents that may be present in a substance – as contaminants, as by-products of production, or arise during processing or storage of a substance, for example:
- residual solvents
- process-related impurities arising from the manufacturing process
- incidental metals and non-metals, for example: lead, arsenic, selenium
- agricultural and veterinary chemicals, for example: pesticides, fumigants
- general contaminants, for example: dioxins, polychlorinated biphenyls
- manufacturing by-products, for example: reagents, catalysts, co-extractives
- degradation products
- radionuclides – particularly where substances might be sourced from contaminated areas
- radiolytic residues
- microbial contamination
- mycotoxins, for example: aflatoxins, ochratoxin A
The presence of impurities and incidental constituents should be minimised consistent with legal and appropriate production, processing and storage practices, for example: principles of Hazard Analogy Critical Control Point or Good Manufacturing Practice. Reliance on finished-product testing alone is not a comprehensive means of managing their presence.
The potential for the manufacturing process to concentrate residues should be addressed. A summary should be provided of any degradation studies carried out to identify impurities arising from exposure to stress conditions, for example: heat, light, pH or moisture.
The draft compositional guideline must include requirements for all known or likely impurities and incidental constituents.
Specifications and descriptions of analytical procedures must be submitted. As a starting point, the tests or methods used in pharmacopoeial references should be used. Other useful references include the methods used by the US Environmental Protection Agency (US EPA) and the US Food and Drug Administration (FDA).
Where non-compendial methods are used, appropriate validation should be provided. Analytical procedures should be validated in accordance with the scientific guideline ICH Q2(R1): Note for guidance on validation of analytical procedures: Text and Methodology (CPMP/ICH/381/95).
Chemical substances and substances of animal origin
Provide information for residual solvents, organic or inorganic impurities or toxins, and any ingredient related impurities in line with guidance ICH Q3A(R2) and ICH Q3C(R8).
Herbal material (herbal substance and herbal extract)
Provide information for organic or inorganic impurities or toxins, microbial contaminants, heavy metals, pesticide residues, and mycotoxins in line with section 5.1 of EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3.
Microorganisms
Provide information as described under heading ‘Impurities and incidental constituents’ of ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’.
Residual solvents
Any solvent(s) that may be used in the production, preparation, manufacturing or formulation should be controlled as per the requirements of the BP supplementary chapter for ‘Residual Solvents’. Specifically, address solvents that are included in the Ph. Eur. general texts 5.4‘SC IV D. Residual solvents’, and Ph Eur method 2.4.24 ‘Appendix VIII L, Residual solvents’.
Elemental impurities
The material should comply with default standard limit tests for heavy metals, for example: lead, cadmium, mercury and arsenic.
The Poisons Standard may stipulate a particular limit for a metal or non-metal constituent in a substance, for example: a substance containing more than 10 mg/kg lead would be subject to the conditions of the Poisons Standard. If a substance is subject to the conditions of a Schedule (or applicable Appendix) to the Poisons Standard, then it is not acceptable as a permitted ingredient for use in listed medicines.
If the Poisons Standard requirements are not applicable, concentration limits for elemental impurities can be determined using the concept described under Option 1 of the ICH Q3D(R1) guideline1 This method is adopted for determining concentrations in drug substances for oral and inhalation routes of administration. Option 1 calculates a common permissible target elemental concentration for each component in the drug product with daily intakes of not more than 10 grams. The equation for calculating the permitted concentration of an elemental impurity is as follows:
Concentration (µg/g) = PDE2 (µg/day) / daily amount of drug product (g/day)
The permitted concentrations of elemental impurities for Option 1 are presented in Appendix 2, Table A.2.2 and apply for drug products, drug substances and excipients in the ICH Q3D(R1) guideline.
Substance for other routes of administration other than oral and inhalation
When PDEs are necessary for other routes of administration, PDEs may be derived from an established PDEs. According to ICH Q3D(R1) guideline, elemental concentration for other routes of administration (e.g. dermal) may be determined by applying a correction factor to an established PDE. For example, when no local effects are expected, if the oral bioavailability of an element is 50% and the bioavailability of an element by the intended route is 10%, a correction factor of 5 may be required.
Herbal material
Active herbal materials and excipients used must comply with the requirements of the relevant monographs, e.g: Substances for pharmaceutical use (2034), Essential oils (2098), Herbal drug extracts (0765), Herbal drugs (1433), Herbal drug preparations (1434).
Pesticide residues and environmental contaminants (including agricultural and veterinary substances)
Pesticide residues may be found in a raw material (e.g. herbal material) as a result of intentional treatment or from inadvertent environmental contamination, of particular relevance are:
- organochlorins (for example: dichlorodiphenyltrichloroethane and endosulfan)
- organophosphates (for example: chlorpyrifos and parathion)
- carbamates (for example: carbaryl and methomyl)
The effects of processing and storage may affect these residues and result in a concentration or reduction of residues in finished goods.
The method, acceptance criteria, methodology and limits stipulated for pesticide residues in the default standards, for example: BP Appendix XI L – ‘Pesticide Residues’, should be followed as well as any additional residue limits that may be relevant. If a medicine substance contains a pesticide residue that is not specifically restricted in the BP, then the risk associated with that pesticide should be assessed based on the generic approach described in the BP. Applicants should identify the likely pesticide residue risks; determine the likelihood and consequences of these risks; and develop, implement and review approaches for managing these risks.
Information from the US Environmental Protection Agency or the Codex Committee on Pesticide Residues can often provide good information about the effects of processing for specific chemicals. Other sources of information include pesticide manufacturers.
Other organic or inorganic impurities or toxins
Other organic or inorganic impurities or toxins may include:
- foreign matter
- total ash
- sulfated ash/residue on ignition
- ash insoluble in hydrochloric acid
- related substances, for example: synthetic impurities, degradation products
- other manufacturing by-products, for example: reagents, catalyst residues or process impurities
- radionuclides: where substances are sourced from contaminated areas
- radiolytic residues: for substances sterilised using ionising radiation
- residues of decontamination treatments
- any other organic or inorganic impurities or toxins (for example: dioxins, polychlorinated biphenyls and microbial toxins such as aflatoxins, ochratoxins)
The likely presence of manufacturing by-products (for example: catalyst residues, synthesis or process impurities and degradation products) should be determined and typical levels in regular production batches documented, particularly where they are of significance to safety or quality. Attention should also be given to the presence of isomers, metabolites and coextractives.
Substances may be sterilised using ionising radiation. You should consider what radiolytic products may be formed in the substance and what components of the substance may be affected by such treatment, for example: vitamin A. You should have documentation about substances that have been irradiated, monitor levels of radiolytic products or components and, if necessary, establish and document limits.
If a decontaminating treatment has been used, it must be demonstrated that the quality of the substance has not been adversely affected and that no harmful residues remain. In relation to other pharmaceutical raw materials and finished products, it is recommended that ethylene oxide be used only when essential and where alternative processes and/or decontamination agents cannot be used. Refer to the scientific guideline: 'Note for Guidance on Limitations to the use of ethylene oxide in the manufacture of medicinal products’. CPMP/QWP/159/01. In relation to herbal materials, the BP dictates that ‘the use of ethylene oxide for the decontamination of herbal products is prohibited’.
Depending upon the substance, specific contaminants (for example: dioxins and polychlorinated biphenyls) may be present and the range of their concentrations should be given.
Microbial contaminants
Microbial limit testing is an important parameter for quality assurance of a substance. Where applicable, there may be a need to specify the total count of aerobic micro-organisms, the total count of yeasts and moulds and the absence of specific objectionable bacteria as per Appendix XVI D. Microbiological Quality of Non-sterile Pharmaceutical Preparations and Substances for Pharmaceutical Use, Ph. Eur. general text 5.1.4. Microbial counts should be determined using pharmacopoeial procedures or other validated procedures. The source of material should be taken into account when considering the inclusion of possible pathogens (e.g. Campylobacter and Listeria species).
The Therapeutic Goods Order No. 100 'Microbiological Standards for Medicines' mandates that any finished product that contains the ingredient, alone or in combination with other ingredients, must comply with the microbial acceptance criteria set by Clause 11 of the Order. While substance manufacturers are encouraged to include limits for objectionable microorganisms, it is the product into which those substances are formulated that is subject to a legally binding set of criteria.
Substances of animal origin
Provide tests/methods for specified microorganisms as per Appendix XVI B. Microbiological examination of non-sterile products, Ph. Eur. general chapter 2.6.13.
Herbal materials (unprocessed herbal substance and herbal extracts)
Provide microbial enumeration tests and test for specified microorganisms as per Appendix XVI F. Microbiological examination of herbal medicinal products for oral use and extracts used in their preparation, Ph. Eur. Method 2.6.31.
Microorganisms
Provide information as described under heading ‘Impurities and incidental constituents’ of ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines'.
Reference standard
Authentication of reference materials
The following scientific guidelines are applicable:
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96
- Guideline on Specifications: Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3.
- ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products CPMP/ICH/365/96
- Guide to good manufacturing practice for medicinal products Part II PE 009-16
A reference standard is a reference material prepared for use as the standard in tests, such as identification, assay and impurities testing. Information should also be provided about how these reference substances were established, and where applicable, how their potencies were assigned. Where ‘in-house’ reference materials are used, appropriate testing should be performed to establish fully the identity and purity.
Herbal material
Provide reference standard(s) in line with guidance section 3.7 of EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3.
Chemical substance and substance of animal origin
Provide reference standard(s) in line with section 2.11 of CPMP/ICH/367/96
Chemically derived excipients for dermal use only
A reference standard is not required however the manufacturer should ensure that the reference material is traceable to a verified standard.
Microorganisms
Provide reference standards in line with guidance under heading ‘Reference standard’ of ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’.
Specifications
You must include information on the specifications used to ensure the quality of the substance. A specification is a list of tests, references to analytical procedures, and appropriate acceptance criteria which are numerical limits, ranges, or other criteria for the tests described. It establishes the set of criteria to which a substance should conform to be considered acceptable for its intended use. This information will be included in a compositional guideline.
The following scientific guidelines provide guidance on specifications:
- Guideline on Specifications: Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96
- ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/biological products CPMP/ICH/365/96
- ICH Q2(R1) Validation of analytical procedures: text and methodology CPMP/ICH/381/95
- Guideline on Excipients in the Dossier for Application for Marketing Authorisation of a Medicinal Product EMEA/CHMP/QWP/396951/2006
Compositional guideline
You must provide specifications in the form of a compositional guideline using the compositional guideline templates. The information in the compositional guideline will be derived from the information you provide during the application such as the description, characterisation (identity, assay, impurities) analytical methods and acceptance criteria. The headings in the compositional guideline template correspond to headings within the ‘Information required to demonstrate QUALITY for substances not subject to a monograph in a default standard’ section of this guidance document.
The method of analysis used to establish compliance with the limits must be included in the compositional guideline, for example: high-performance liquid chromatography (HPLC). Methods in pharmacopoeias for similar substances should be used wherever possible, for example: pH measurements. If the method and limits are based on a pharmacopoeia or published reference, these references must be provided. If proprietary or non-compendial methods are employed, a brief description in the draft compositional guideline is acceptable, for example: ‘HPLC: Column: Silica (250 mm x 4.6 mm, 5 µm), Mobile phase: ACN/water (4:1); Flow rate: 1.8 mL/min; Column temperature 35o C, RI Detection’. However, you must provide validation data of the analytical methods referred to in the compositional guideline in accordance with ICH Q2(R1) if the tests used are not pharmacopeial methods. Any methods or procedures identified in the compositional guideline should be able to be reproduced by an independent authority.
In general, the information included in a compositional guideline should:
- provide the physical and chemical properties of the substance
- identify and quantify major components and any significant (that may affect the safety or quality of the substance) minor components
- distinguish the substance from similar substances, adulterants or substitutes
- be specific for components of safety and/or therapeutic significance
- provide the limits of possible contaminants and impurities
- describe the biological, botanical, chemical and physical variations that may reasonably occur between batches of the substance
- be capable of providing for objective validation of the substance’s composition using described analytical methodology
If certain parameters included in the compositional guideline template are not relevant, these can be omitted provided that justification is given, for example: ‘The production of this substance does not require the use of solvents and therefore the compositional guideline requirement for solvent residues has been omitted’.
Where there are external quality references for a substance such as the Joint FAO/WHO Expert Committee on Food Additives (JECFA) monograph that provide relevant information to address the required tests outlined in the compositional guideline template; applicants can use the information from these references and include them in the compositional guideline template and update methods, criteria or specifications as appropriate.
For further information about compositional guidelines, see Overview of Compositional Guideline.
Complex substances
Complex substances, particularly those of herbal origin, may be subject to variation due to such factors as: genetic variation; geographic variation; growing conditions; maturity and time of harvesting; post-harvest treatment; storage conditions; and/or processing treatments. Limits taking into account this variation must be included in the compositional guideline and justification for the limits provided, for example: ‘the components in certain plants may vary seasonally and batches may contain, at certain times of the year, less of a certain component’.
Excipients for dermal use only
Provide a CG with a description, general properties, identity and impurities. An assay is not a requirement when the excipient is only to be used in a formulation for its physical properties (e.g. emulsifier, thickener). Please note that a restriction may be placed so that the excipient will only be limited to use for physical properties in line with the purposes for which the substance was evaluated for.
Batch analysis
A description of batches and batch analyses results must be provided.
The date of manufacture, batch size and number, place of manufacture, analytical methods used, should be provided. Test results (e.g. impurity levels) should be expressed numerically. Results that merely state that the material ‘complies’ with the test are insufficient.
If available, provide certificates of analysis for any batches of material used in toxicity tests and clinical trials reported in support of the application. This will assist to determine if the substance intended for supply is the same as that for which safety has been provided.
The following scientific guidelines provide guidance on batch analysis:
- ICH Q3A(R2): Impurities in new drug substances CPMP/ICH/2737/99
- ICH Q3C(R8) Impurities: guideline for residual solvents EMA/CHMP/ICH/82260/2006
- ICH Q6A Specifications: Test procedures and acceptance criteria for new drug substances and new drug products: chemical substances CPMP/ICH/367/96
- ICH Q6B Specifications: Test procedures and acceptance criteria for biotechnological/ biological products CPMP/ICH/365/96
- Guideline on Specifications: Test procedures and acceptance criteria for herbal substances, herbal preparations and herbal medicinal products/traditional herbal medicinal products EMA/HMPC/CHMP/CVMP/162241/20051 Rev. 3
Active ingredients
For ingredients intended for use as an active, provide certificates of analysis for at least two, commercial-scale production batches to demonstrate routine compliance with the proposed compositional guideline. If data on commercial-scale batches is not available, provide certificates of analysis for three primary batches to a minimum of pilot-scale manufactured by the same process as intended for commercial-scale batches.
Excipient ingredients
For substances intended only for use as an excipient only, a certificate of analysis for at least one commercial batch or two pilot scale batches to demonstrate routine compliance with the proposed compositional guideline must be provided.
Stability test
The following scientific guidelines provide guidance on stability studies:
- ICH Q1A(R2) Stability testing of new drug substances and products CPMP/ICH/2736/99
- Stability testing of existing active substances and related finished products CPMP/QWP/122/02, rev 1 corr
- Guideline on quality of herbal medicinal products/ traditional herbal medicinal products EMA/HMPC/201116/2005 Rev. 2
- Guideline on excipients in the dossier for application for marketing authorisation of a medicinal product EMEA/CHMP/QWP/396951/2006
The purpose of stability testing is to provide evidence on how the quality of a substance varies with time under the influence of a variety of factors such as temperature, humidity and light to establish a re-test period. Stability studies aim to determine the quality of a substance by assessing the parameters within the compositional guideline that are susceptible to change during storage and are likely to influence the quality, safety and efficacy of a substance. Some substances produce degradants during storage, so the purpose of stability studies is also to identify these and ensure there are no consequential safety concerns. For example, marine oils can produce peroxides, and these are generally identified in stability studies and need to be effectively controlled. As such, the stability testing should demonstrate that the criteria and limits that correspond to the quality of the substance do not change during storage period and there are no degradants of safety concern.
Stability testing should be conducted in accordance with the TGA-adopted scientific guidelines ICH Q1A(R2) and CPMP/QWP/122/02, rev1 corr. For all substances (excluding excipients for dermal use only), real-time and accelerated stability data must be provided for two commercial scale batches or three pilot scale batches.
The pilot scale batches should be manufactured by the same process as intended for commercial-scale batches. For all new/ novel drug substances, real-time stability data must be provided for minimum 12 months, and accelerated stability data for 6 months as per the guideline ICH Q1A(R2).
For all existing active substances (that have been authorised previously through a finished product within the European Community), real-time stability data must be provided for minimum 6 months, and accelerated stability for 6 months as per the guideline CPMP/QWP/122/02, rev1 corr.
It is possible to provide stability data for a reduced number of batches or batch sizes if sufficient scientific sound evidence is provided to demonstrate that the substance is stable and that no degradants of concern are generated/produced during storage (e.g. some minerals that are known to be chemically stable and have published literature to support this).
The application should include a summary of the studies undertaken (conditions, batches, analytical procedures) as well as a brief discussion of the results, conclusions and the proposed re-test date where relevant.
A tabulated summary of the stability results, with graphical representation where appropriate should be provided.
Stress testing
Stability testing is not required if stress testing data can be provided that demonstrates the absence of degradants (refer to section 2.1.2 of the ICH Q1A(R2) and CPMP/QWP/122/02, rev1 corr.
Excipients for dermal use only
Stability studies are not required for excipients for dermal use only
Information required to demonstrate SAFETY
The safety of a substance for use in listed medicines may be supported by history of use, published literature and/or original study data. A combination of information from human exposure information and in vivo and in vitro nonclinical studies can be used to address the toxicological profile information requirements. Clinical and other efficacy information, while not evaluated from an efficacy perspective, often include information on adverse events that is useful in the safety evaluation.
The two factors that determine the overall risk associated with a substance are the level of exposure to the substance, and the hazard (its actual or potential to cause harm) that is intrinsic to that substance. The information to be provided in the sections History and pattern of previous human use and Biological activity allows assessment of the level of consumer exposure to the substance, both at the existing levels and usages, and at the proposed dosage in the application. The information to be provided in the Toxicological data section allows assessment of the hazard presented by the substance for the proposed route(s) of administration.
The safety profile of a substance for use in listed medicines must be consistent with the low-risk status of these self-selected medicines. Conditions may be placed on the use of an ingredient to ensure an appropriate level of risk if it is determined that the hazard posed by the proposed use is beyond that considered acceptable for listed medicines. For example: label advisory statements or restrictions to maximum daily dosages commensurate with exposure data may be required.
When multiple routes of administration are proposed (e.g. oral and topical use) in the application, then safety data should be provided to address the use of the substance for all proposed routes of administration. When a route of administration that may have effects on multiple bodily systems (e.g. dental administration – both mucosal and oral absorption need to be considered), then data should be supplied to address the likely exposure from all relevant routes of administration.
Although different salts may have slight differences in chemical structure, they have the potential to cause different physiological effects. For example, different magnesium salts, when administered orally, have different risks of causing diarrhoea. Unless similarity in the pharmacology (i.e. generation of similar degradation and metabolites that have no hazardous effects on the host, demonstrated using in vitro and in vivo studies) between salts is established, individual salts are considered distinct substances for the purpose of the safety evaluation. Biological equivalence must be established between different salts so that toxicological data generated by another salt can be leveraged to justify the lack of data for the salt under evaluation.
Key aspects of the safety dossier include:
- Relevant information based on a comprehensive literature search including both positive and negative reports.
- Published material such as papers, expert reports and reviews must be provided in full. Copies of unpublished study reports must also be provided, if available. Abstracts are not acceptable as evaluable material.
- Ideally, pre-clinical studies should be performed in laboratories that hold GLP or accreditation to ISO/IEC 17025 or equivalent, e.g. NATA, United Kingdom Accreditation Service, American Association for Laboratory Accreditation. Clinical trials should be undertaken in accordance with Good Clinical Practice or generally accepted scientific standards The report should include certification of compliance in the conduct of each study.
- Safety Data Sheets (SDS) alone are not acceptable.
For a summary of core information requirement for different substance types, see Appendix (Table 34).
Microorganisms
References to a supplementary guidance document ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’ are made under the relevant sections of this document, and are intended to complement this document. The supplementary guidance applies to microorganisms defined as whole and intact cells of bacteria and fungi (including yeasts) that are live or non-viable.
When a substance consists of non-viable microorganisms whose membrane integrity has been compromised such that the cells are no longer whole/intact, it is assessed like most other substances. Information required to demonstrate the safety of such substances is outlined in this document.
Substances for dermal use
All substances for ‘dermal use only’ need to address the requirements under the following safety headings:
- Systematic literature search
- History and pattern of human use
- Adverse reactions
- Substances of human or animal origin
There are different requirements if a substance is not absorbed beyond the superficial barrier of the epidermis (the stratum corneum). The stratum corneum is the rate-determining layer when considering the absorption of substances through the skin. Absorption studies are required for all substances in products intended for dermal use unless they are highly unlikely to be dermally absorbed. If you are not providing absorption studies, and the substance is highly unlikely to be dermally absorbed, you must provide scientific justification with full text references, discussing the relevant factors influencing dermal absorption for that substance:
- molecular weight
- charge
- lipophilicity
- whether the substance is degraded to small molecule moieties after skin application (e.g. metabolites generated by skin enzymes or sun exposure)
- duration of exposure
- the dosage
If a substance does not demonstrate absorption beyond the stratum corneum and does not biologically or chemically react with the skin, then limited safety data is required. Figure 1 can be used to demonstrate this.
If a substance is demonstrated to not biologically or chemically react with the skin but may contact other tissues such as leading to oral exposure or inhalation (e.g. substances applied on the face, substances that do not have requirements to wash hands after use, sprays), provide a minimum in silico analysis for mutagenicity (or genotoxicity study if available) to address lack of genotoxicity. If the in silico analysis for mutagenicity is positive for the substance, then the information required will be similar to other substances that are systemically absorbed. An in silico analysis for mutagenicity may not be necessary for some substances (e.g. approved food additives).
If absorption has been demonstrated (or cannot be excluded), then the information required will be similar to other substances that are systemically absorbed.
Figure 1: How to demonstrate if a substance is not absorbed beyond the stratum corneum and does not react with the skin.
* Note that if local tolerance studies have not been undertaken in broken skin or in the eye, respective restrictions ‘not to be included in medicines intended for use on broken skin’ (or words to that effect) or ‘not to be included in medicines intended for use in the eye’ (or words to that effect) will apply to the proposed substance.
See description of image to access hyperlinks in the image.
{kind=link}
The image shows a flowchart diagram describing absorption tests and required information for substances.
The top box is labeled "Absorption" and contains the following points:
- In vitro (OECD 428) and in vivo (OECD 427) percutaneous absorption tests, OR
- Human absorption studies/human bioavailability data, OR
- Information to address factors influencing dermal absorption
Below this, the diagram branches into two paths: "Absorbed (or cannot be excluded)" and "Not absorbed".
The "Absorbed" path leads to a box labeled "Information required" with two main sections:
- Biological activity
- Pharmacokinetics (Distribution, Metabolism, Excretion)
- Pharmacodynamics (Primary pharmacodynamics, Safety pharmacology, Known drug interactions)
- Toxicological data
- Using acute studies (if available), repeat-dosed studies, clinical trials and/or alternative studies to address endpoints including:
- Maximum daily dose
- Duration of use
- Genotoxicity
- Carcinogenicity (if for >6 mth of continuous use)
- Reproductive and developmental toxicity (if for use during pregnancy, lactation and/or in paediatric population)
- Local tolerance
- Using acute studies (if available), repeat-dosed studies, clinical trials and/or alternative studies to address endpoints including:
The "Not absorbed" path leads to a box labeled "Local tolerance" containing:
- Skin irritation study: OECD 404 (acute dermal irritation), OECD 439 (in vitro skin irritation) or human repeat patch insult test (HRPIT)*
- Skin sensitisation study: OECD 406 or HRPIT*
- Eye irritation study: OECD 405 (acute eye irritation) or OECD 492 (reconstructed human cornea-like epithelium)*
- Phototoxicity studies (phototoxicity and photosensitisation) if intended for dermal use where there is potential for sun exposure
* Asterisks indicate footnotes in the original image.
Systematic literature search
The dossier must include a well-constructed (systematic) literature search with the search strategy. For more information, refer to Literature-based submissions for listed medicines and registered complementary medicines. A literature search should include all information that is available on a reliable search engine such as Medline and/or Embase to enable an unbiased coverage of publicly-available information. The information submitted should be relevant to the particular substance and reflect the totality (balance and range) of the available evidence. Consistent evidence from different studies increases the strength of the evidence. All evidence, both favourable and unfavourable, should be documented. Where there is a large search output, it may not be appropriate to include all of the papers in the submission and in this case, justification for the inclusion/exclusion of papers should be provided, for example: on the basis of the quality of the study or provision of a recent review of high quality.
History and pattern of human use
The information in this section should determine the existing exposure of the consumer population to the substance.
To establish safety, sufficient numbers of people must have been treated or otherwise exposed to the substance or to products containing the substance (or to a substance justified as essentially identical to the substance in question) over a considerable period of time.
When there is sufficient clinical and/or historical human evidence to support safety of a substance for specific route(s) of administration at the proposed dose and duration of use, and in the proposed population, conventional toxicological studies involving animal and in vitro studies may not be necessary. However, where human evidence is lacking or there are clear reasons to suspect that clinical data are deficient or incomplete, the safety assessment will need to be supported by other studies e.g. repeat-dose toxicity.
Some substances are available in countries that have regulatory controls that are different to Australia’s regulation of listed medicines. For example, some listed medicine products are regulated as foods or dietary supplements in other countries. Generally, food regulation does not include pre-market evaluation, rigorous post-market pharmacovigilance or a system for adverse reaction reporting. The use of substances regulated under less stringent controls may not provide a high degree of assurance of their safety in use, particularly if there is limited control on composition and adverse reaction reporting. However, information about such use may still be helpful in supporting safety.
Use in therapeutic goods (Australian and International)
Where a substance has been an ingredient of a registered medicine, either in Australia or internationally, such history of use and its eligibility for use in listed medicines will be considered, but it is essential to demonstrate that the proposed substance is the same as that used in those goods.
Post-marketing experience with other products containing the same or a similar substance should be supplied in the application. Post-marketing experience can be provided from use of the substance in jurisdictions other than Australia.
Provide information on the availability of the substance in other countries, the period of time it has been available, and the regulatory conditions controlling its availability.
Provide reports from international regulatory authorities or agencies and discuss these if available, for example:
- the Joint Food and Agriculture Organization/World Health Organization Expert Committee on Food Additives (JECFA)
- the US Food and Drug Administration (FDA)
- European Food Safety Authority (EFSA)
It is important to highlight the purpose of the particular agency’s evaluation, which may have been for a more restricted purpose than that proposed in your application e.g. an evaluation of safety for cosmetic use (dermal) is unlikely to have considered safety for oral use. Similarly, an evaluation of a food additive is unlikely to have considered dermal toxicity. These reports may also have recommended particular restrictions on the substance; if so, these should be discussed. Provide the outcome of such applications. Applicants must not omit any scientific or regulatory report that could influence assessment of safety of the substance.
Use in food
Information from well-established use of the substance in food can be used to support or establish safety. ‘Well-established use’ means that a sufficient number of people were exposed to the substance over a period sufficient to support the safety of the substance for its intended purpose. Usually a substance that has a long history of use will have information published in official pharmacopoeias and other published literature. However, in some cases, particularly when well-established use cannot be demonstrated based on written records (e.g. indigenous use), you should submit information gathered from traditional users.
Where results from epidemiological studies of food or dietary supplements are of sufficient power or other adequate post-market safety studies are available, this may be sufficient to support safety.
If the use of a substance is permitted in food in Australia, any applicable reference in the Australia New Zealand Food Standards Code should be given.
For substances that may be naturally found in food, documented evidence of the presence of these substances in general human consumption at similar levels should be provided. It is not sufficient to include only evidence of short-term consumption by a particular sub-group of the population or limited consumption.
A substance used in therapeutic goods may present a different risk profile to that resulting from its use in food. Other components in the food matrix, such as fibre, may affect the rate of absorption or otherwise interact with the substance when it is present in food. There may be no such effect when the substance is delivered in a therapeutic formulation. These matrix effects may be significant in terms of safety for some substances and may require limits on the proposed maximum single or daily dose.
Traditional use
If you are relying, in part or in total, on evidence of traditional use to demonstrate safety, you must clearly indicate whether the substance under review is the same as that used traditionally. That is: the same plant part, method of preparation (e.g. aqueous or ethanolic extraction), dosage and dosage form, route of administration and typical schedule of administration.
The population and culture in which this tradition occurred must be identified. In some cases, evidence of traditional use, for example, aboriginal bush remedies would require robust anthropological research data.
Modern extraction methods or other processes may produce, in some cases, substances that have a considerably different compositional profile from those produced using traditional methodology. It is insufficient to rely entirely on evidence of traditional use to support the safety of these substances. For example, modern highly concentrated Actea racemosa (black cohosh) herbal extracts have been associated with serious adverse reactions that have not been reported for traditional extracts. In some instances, the extraction of a substance from its natural matrix may make it more prone to oxidation to a toxic product or to inactivation, for example: carotenoids or resveratrol.
History of safe use
Many substances have been in use in other countries in cosmetic formulations for a substantial length of time, and this provides some assurance of safe use in a human context. Evidence of the inclusion of a substance in other formulations should be provided, along with information on how the existing use is relevant to use in listed medicines. Discussion should address the length of time the substance has been in use, the level of human exposure from the existing use, the frequency of use in the current setting, and what existing safety monitoring or mechanisms to detect safety signals are available to provide an assurance of safety.
Some substances will only have been used in certain types of formulation (e.g. oil-based ointments) and discussion or further information in Biological activity and Toxicological data. should be provided to establish the safety of the substance in other formulation types.
Applications relying on history of safe use may wish to consider whether a substance is naturally found in the diet (refer to ‘Use in food’) or endogenously present in the human body. For endogenous substances present in the human body, a discussion of the existing amounts should be supported using robust and reliable literature. The discussion should consider the bioavailability of the substance and how this compares to the endogenous amounts found in an average adult. Discussion should also be provided as to the expected biological effects from consuming the substance in addition to the diet or endogenous levels.
Microorganisms
Provide information as per heading ‘History of safe use’ in ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’.
Summary of overall human exposure from all sources
To assess the safety of a substance for use in listed medicines, it is necessary to estimate the overall human exposure to the substance, particularly if the substance is present in a typical diet. The exposure evaluation determines the amount of the substance that populations may be exposed to from all sources.
In determining possible total exposure to a substance, consideration must be given to the net and total amount of exposure from other sources and from use in listed medicines. The duration and route of exposure must be considered.
Where possible, information on population exposure data should be included in the application. Where data are not available on the particular substance, data derived from related substances (such as the precursor, metabolite or a close analogue) may be useful as supporting evidence. For some nutrients and food types, the National Nutrition Survey will contain useful estimates of consumption.
For substances proposed for dermal use, appropriate dermal exposure models (external and internal) outlined in Notes of guidance for the testing of cosmetic ingredients and their safety evaluation, 12th revision (SCCS/1647/22) should be used to calculate the maximum daily exposure based on different exposure scenarios of the substance. Where necessary, exposure of vulnerable populations should be assessed separately. Where a substance does not have any restrictions limiting to adult use only, consideration must be given to account for the differences in skin surface area over body weight ratios between children and adults.
Biological activity
Data in this section should be provided for each route of administration proposed in the application. Consideration should also be given to indirect exposure by other routes of administration, for example: inhalation exposure from a substance that is intended for use as a spray for topical use, or gastrointestinal exposure from a medicine for dental application.
The first section on pharmacokinetics primarily considers whether the substance is absorbed, and the degree of that absorption (i.e. whether systemically absorbed or remains on the administration site). The second section on pharmacodynamics considers substances that are systemically absorbed.
All studies should be conducted using internationally recognised methodology as described in relevant Organisation for Economic Co-operation and Development (OECD), European Medicines Agency (EMA), International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidelines or other approaches that are equivalent.
Microorganisms
Biological activity of microorganisms is assessed differently from other substances. Provide information as outlined under the heading ‘Biological activity’ in ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’.
Pharmacokinetics
Pharmacokinetic studies describe the mechanisms by which the substance is processed by the host and are crucial in assessing systemic exposure. Appropriate studies, including human exposure and/or animal in vivo and in vitro studies using appropriate experimental models and routes of administration, should be provided on:
- Absorption
- Tissue distribution and storage
- Metabolism
- Mode and extent of excretion or elimination of the parent substance and its degradation and metabolism products.
The key purpose of absorption studies is to determine the extent of systemic exposure to the substance, and to establish the relevance of animal models for human exposure. This exposure determines which types of toxicology data must be considered during evaluation of the safety of the substance. For example, if a substance is intended for dermal application only, and information is available to demonstrate that the substance is not absorbed through the skin, and that any metabolites generated by skin enzymes or sun exposure are also not absorbed, then acute and chronic toxicity information is likely to be unnecessary.
Substances for all other routes of administration other than dermal use
For substances proposed for all routes of administration except dermal, refer to ICH S3A.
Substances for dermal use only
For substances proposed for dermal use, refer to the OECD Guidance document for the conduct of skin absorption. One of the following is required:
- in vitro (OECD 428) and in vivo (OECD 427) percutaneous absorption test, or
- Human absorption studies or human bioavailability data, or
- Information to address factors influencing dermal absorption
If available, an in vivo determination of oral absorption should also be provided to establish systemic exposure to enable interpretation of toxicological data. Otherwise, scientific justification demonstrating the lack of significant acute oral toxicity must be provided. These may be sourced from post-market surveillance data, case reports, poison centre information, or epidemiological studies.
For substances for dermal use on unbroken skin only, that are demonstrated to not be absorbed beyond the stratum corneum, no other pharmacokinetic information is required (Refer to Figure 1).
If systemic absorption has been demonstrated (or not excluded), studies in accordance with ICH S3A guideline (CPMP/ICH/384/95) should also be provided to address tissue distribution and storage, metabolism, and the mode and extent of excretion or elimination of the parent substance and its degradation and metabolism products.
Herbal materials
Provide pharmacokinetic studies of active biocomponents of the substance in accordance with ICH S3A (CPMP/ICH/384/95).
Microorganisms
Provide information as outlined under the heading ‘Pharmacokinetics’ in ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’.
Pharmacodynamics
The objectives of pharmacodynamics studies are two-fold. Firstly, to ascertain how the substance works in or on the human body (mechanism of action) and secondly, to identify undesirable pharmacodynamic properties of substances that may have relevance to its human safety. They also facilitate the evaluation of adverse effects of the substance observed in toxicology or clinical studies and therefore assist in investigating the mechanism of adverse effects observed or suspected. For example, if during evaluation it is determined that the substance demonstrates neurotoxicity, immunotoxicity, or antigenicity at a level of exposure similar to that proposed in the application, it is unlikely that the substance would be considered suitable for use as an ingredient in listed medicines.
Appropriate studies, including human exposure and/or animal in vivo and in vitro studies using appropriate experimental models and routes of administration, should be provided on:
- Primary pharmacodynamics
- Safety pharmacology to study the effects of the substance on the following vital functions:
- Central nervous system
- Cardiovascular system
- Respiratory system
- Known pharmacodynamic drug interactions identified in the literature search
Substances where systemic absorption has been demonstrated (or not excluded)
Information to address the above pharmacodynamic endpoints should be in accordance with scientific guideline ICH S7A (CPMP/ICH/539/00).
Substances for dermal use on unbroken skin only, that are demonstrated to not be absorbed beyond the stratum corneum and do not react with the skin
(Refer to Figure 1).
Information on pharmacodynamics is not required.
Herbal materials
Provide information to address the pharmacodynamic endpoints above for the proposed substance. If these are not available for the proposed substance, pharmacodynamic studies of individual active components of the proposed substance should be provided.
Microorganisms
Provide information as outlined under the heading ‘Pharmacodynamics’ in ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’ .
Toxicological data
Data provided to address the toxicological end-points detailed in this section can be a combination of in vitro studies, animal studies, other suitable alternatives and/or human clinical studies. The core principles of each toxicological end-point defined below should be addressed in all applications. Concentrations and/or amounts of the substance used in all studies must be clearly stated.
Data submitted to address toxicological endpoints for new substances must consider both local and systemic toxic effects. However, if it can be demonstrated that the substance does not move into bodily systems beyond that which the substance is administered to (determined by pharmacokinetics: absorption studies above), and if indirect exposure to another bodily system is unlikely, then consideration of systemic toxic effects is not necessary. For example:
- If a substance is intended for use in medicines for dermal application, if the study data demonstrates that there is no absorption through the skin (i.e. not systemically absorbed), then only local tolerance data is required (Refer to Figure 1).
- If a substance is intended for oral application (e.g. within the mouth), and data demonstrates that there is no absorption from either the gastrointestinal tract or oral mucosa, then in general only repeat-dose toxicity or human chronic exposure, and local tolerance studies would be required.
- If the substance is intended primarily for use for dermal application, but oral exposure is likely to occur (e.g. lip balm), oral exposure must also be considered and toxicological data provided if exposure is significant. If the data demonstrates that there is minimal absorption from either the gastrointestinal tract or oral mucosa, then in general local tolerance data is required.
Data from clinical trials should be submitted under the appropriate toxicological data sections. Clinical safety studies should consider whether the duration, dosage, targeted population and route of administration of the studies are consistent with the proposed use of the substance under evaluation. You should include haematological tests, liver enzyme tests, electrolytes and urinalysis as clinical endpoints to address clinical safety. Tolerability issues and adverse events arising in those clinical trials in your application should also be addressed. Clinical trial data submitted in support of the safety of a substance will not be evaluated for efficacy.
It is acknowledged that it can be challenging to source conventional toxicity data for listed medicine substances. However, the absence of this data does not imply that the substance is safe. In the absence of this data, it is difficult to complete a safety risk assessment.
Justification to demonstrate why an acceptable level of safety can be assured, even though some studies are lacking, must be provided and may be associated with a history of safe use in other types of product. A substance is not likely to be considered suitable for use as an ingredient in listed medicines if there is insufficient evidence to provide assurance of safety.
Toxicological study details should include the:
- reference to the applicable test guideline
- route of administration
- dose levels, purity and batch details of the test substance
- number of animals or subjects per dose level
- animals’ or subjects’ origin, sex, weight range and age
- frequency at which observations were made
- duration of each study
- the relationship between the time of administration and the onset of the effects observed
- all measurements made
- key outcomes (e.g. established LD50, No Observable Adverse Effect Level [NOAEL], nonmutagenic, non-carcinogenic, biochemical analysis unchanged between test and placebo).
All studies should be conducted using internationally recognised methodology as described in relevant OECD, EMA, ICH guidelines or other approaches that are equivalent.
Further information is available from OECD Guidelines for the testing of chemicals, ICH M3(R2) (EMA/CPMP/ICH/286/1995) and ICH E3 (CPMP/ICH/137/95) guidelines.
Pivotal studies, from which a NOAEL is established, should be undertaken with the substance proposed for use or a substance of comparable composition (subject to justification) to that proposed for use. It should be noted that an Acceptable Daily Intake (ADI) is derived by applying a default safety factor (usually 100x to account for inter-species and intra-species differences) to the NOAEL identified in a repeat-dose toxicity study or carcinogenicity study. The Margin of Safety (MoS) is then derived as a ratio of the Expected Daily Intake (EDI) and the ADI; a value of < 1 is generally considered safe from a toxicological perspective.
A table providing a summary that concisely describes every aspect of toxicity studied should be provided in the application. The summary should identify all substance-related biochemical and physical changes observed in the study, with appropriate cross-referencing to the reports in the main submission. A summary table (see Table 12) should be provided for the clinical trials included in the application.
Substances for dermal use where systemic absorption has been demonstrated (or not excluded)
Calculations of the Systemic Exposure Dose (SED) of the substance can be derived for the proposed amount of the substance using either of the 2 options specified in Notes of guidance for the testing of cosmetic ingredients and their safety evaluation, 11th revision (SCCS/1628/21). A MoS is then calculated as a ratio of the NOAEL (usually derived from a repeat-dose oral toxicity study) and the SED; a value of >100 is considered safe, according to the SCCS Notes of guidance.
Microorganisms
Refer to the heading ‘Toxicological data’ in ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’ for further information. Information addressing genotoxicity is not required. However, all other toxicological endpoints described under the sub-headings below should be addressed.
Table 2: Example format - Summary of toxicological data
| Type of study (reference) Test guideline | Test substance | Animal/Subject details | Treatment details (dose, duration, route) | Measurements | Observations | Key outcomes |
|---|---|---|---|---|---|---|
| E.g. 90-day repeat-dose oral toxicity study in rodents (Smith et al. 2000) e.g. OECD principles of GLP, OECD 408 | Substance (batch number LM1234, 99.5% purity), constituted in grapeseed oil | Sprague Dawley rats (n=10/group/sex) |
| e.g. body weight, organ weight, relative organ weight, blood serum levels, clinical and behavioural observations, histopathology. | e.g. No mortality in all groups. No differences between test and placebo for blood serum levels. | e.g. Diarrhoea symptoms in 1/10 rats in Group 2 considered unrelated to treatment, established NOAEL: 10 mg/kg bw/day. |
| E.g. Doubleblinded, parallel, singlesite, randomised controlled trial (Citizen et al. 2000) e.g. GCP | Substance (batch number LM1234, 99.5% purity), capsule | 107 patients, mean age 56 years: 50 male; 57 female. Overweight but healthy otherwise | 10 mg per day oral for 12 weeks, and 4-week follow up | e.g. blood serum levels, expected duration of use in usual setting, adverse effects, body weight | No differences between test and placebo for blood serum levels. Adverse effects: gastrointestinal symptoms (2/54), rash (4/54), oedema (1/54). | e.g. Safe and tolerable in test group, resolution of adverse reactions in 7/7 patients, no withdrawals despite adverse reactions, similar numbers of adverse reactions in test and placebo groups. |
Acronyms used:
- GLP: Good Laboratory Practice
- GCP: Good Clinical Practice
- NOAEL: No Observable Adverse Effect Level
Toxicological data must be presented under the sub-headings provided below.
Maximum daily dosage
Data must be provided to establish that the maximum daily dose proposed in the application does not present any safety concerns. This data can be acute toxicity studies in animals, repeat-dose toxicity studies, or human clinical trials performed using a dose equivalent to or higher than the proposed dose.
The inclusion of the results of acute dose oral, dermal or inhalation toxicity (LD50 / LC50) testing are not mandatory. These studies should be provided if available. The availability of acute toxicity studies for a novel ingredient may be limited due to international agreement to limit such studies, particularly when data for a similar substance or a class of chemicals are available.
- For acute oral toxicity, refer to OECD 420.
- For acute dermal toxicity, refer to OECD 402.
- For acute inhalational toxicity, refer to OECD 403.
- For repeat-dose oral toxicity, refer to OECD 407 (28-day rodent) and OECD 408 (90-day rodent).
- For chronic oral toxicity, refer to OECD 452 (chronic rodent) and OECD 453 (combined chronic/carcinogenicity rodent).
- For repeated dose dermal toxicity, refer to OECD TG 410 (21/28-day rat/rabbit/guinea pig).
- For human clinical studies, refer to ICH E3 guideline.
Duration of use
Data must be provided to establish the safety of the ongoing use of the substance at the proposed duration of use. This data can be in the form of repeat-dose animal studies, human clinical trials of sufficient duration, or potentially from a detailed history of safe use in an equivalent population.
The objective of repeat-dose studies (sub-acute, sub-chronic and chronic toxicity) is to determine the effects of a substance on a mammalian species following prolonged and repeated exposure. The duration of the repeat-dose study should at a minimum, be related to the duration of the proposed therapeutic use of the substance.
The combined dataset of animal studies should meet ICH M3(R2) guideline which requires repeat-dose data for at least one rodent and one non-rodent mammalian species. Generally, short-term use (up to a week) would need to be supported by a sub-acute, 28-day toxicity study; longer therapeutic use would require a sub-chronic (90 days) study; and continuous use of at least 6 months must be supported by long-term, chronic-exposure studies comprising of at least one rodent study of 6 months duration and one non-rodent study of 9 months duration, in accordance with ICH S4 guideline.
Include detailed results from individual subjects in all toxicity studies and supplementary tables or diagrams, for example: growth curves and tumour incidence tables should be provided. It should be possible to organise tables so that the most appropriate comparisons, for example: control and treated groups, appear on the same page and results of histopathological observations can be readily evaluated in relation to dose, sex and duration of treatment. Provide data in order by species, by route, and by duration from 2-weeks to chronic.
The interpretation of chronic-toxicity studies may be greatly influenced by toxicokinetic considerations, particularly when species differences are apparent. Wherever possible, plasma levels of the test substance (and/or its metabolites) should be measured at steady state.
- For repeated dose oral toxicity, refer to OECD 407 (28-day rodent) and OECD 408 (90-day rodent).
- For chronic oral toxicity, refer to OECD 452 (chronic rodent) and OECD 453 (combined chronic/carcinogenicity rodent).
- For repeated dose dermal toxicity, refer to OECD TG 410 (21/28-day rat/rabbit/guinea pig).
Human clinical trials need to consider the safety profile of a proposed substance over a reasonable duration of time consistent with its intended duration of use. The design of clinical studies can significantly influence the ability to make causality judgements about the relationships between the substance and adverse events. To achieve the objective for adequate characterisation of adverse events over time, the size of the cohort of exposed subjects should be considered. According to ICH E1 guideline, for a substance to be used in a medicine continuously for at least 6 months via oral administration, 100 patients exposed for a minimum of 1 year is considered to be acceptable. The data should be sourced from prospective studies appropriately designed to provide at least one year exposure at dosage levels intended for clinical use. When no serious adverse events are observed in a one-year exposure period this number of patients can provide reasonable assurance that the proposed substance is safe for ongoing use.
Substances where systemic absorption has been demonstrated (or not excluded)
Refer to the following scientific guidelines on general principles for repeat-dose studies:
- ICH S3A – Note for Guidance on Toxicokinetics: The Assessment of Systemic Exposure in Toxicity Studies CPMP/ICH/384/95
- ICH S4 – Note for Guidance on Duration of Chronic Toxicity Testing in Animals (Rodent and Non Rodent Toxicity Testing) CPMP/ICH/300/95
- Guideline on repeated dose toxicity CPMP/SWP/1042/99 Rev 1 Corr
- OECD Guidelines for the Testing of Chemicals
- ICH E1 – Note for Guidance on Population Exposure: The Extent of Population Exposure to Assess Clinical Safety CPMP/ICH/375/95
- ICH E3 – Note for Guidance on Structure and Content of Clinical Study Reports CPMP/ICH/137/95
Genotoxicity
Mutagenicity studies are conducted to determine the potential for a substance to contribute to genetic damage in humans. A basic dossier of genotoxicity studies will generally comprise:
- an investigation of the potential to induce point mutations (base-pair substitution and frame shift) using Ames assays, with and without appropriate metabolic activation system
- an investigation of the potential to induce chromosome damage using mammalian cells in vitro, such as the chromosomal aberration assay, with and without appropriate metabolic activation systems.
If a positive result is returned in either of these two assays, results of the following two in vivo or in vitro tests should be provided:
- an investigation of the potential to induce cytogenetic damage, such as the micronucleus test in the bone marrow or other proliferative cells of intact animals
- an investigation of the potential to induce genotoxic damage involving other than cytogenetic damage (for example: unscheduled DNA synthesis (UDS) or P32 postlabelling adduct formation) and preferably using a tissue known or suspected to be a toxicity target for the substance.
Supplementary tests (for example: sister chromatid exchange) can also be used to provide clarification of unexpected or equivocal results in the basic test package, or to provide additional evidence. In vivo germ cell tests using laboratory animals (for example: mouse-specific locus tests, heritable translocation assay) could also be considered for the evaluation of a suspected mammalian mutagen.
For further information, refer to the following documents:
- ICH S2(R1) – Guideline on genotoxicity testing and data interpretation for pharmaceuticals intended for human use EMA/CHMP/ICH/126642/2008
- OECD Guidelines for the Testing of Chemicals
If reports of many studies are submitted, they should be presented under appropriate subheadings: ‘In vitro’ and ‘In vivo’, both with further subheadings such as ‘Gene mutations’, ‘Chromosomal effects’, ‘Unscheduled DNA synthesis’.
Substances for dermal use on unbroken skin only, that are demonstrated to not be absorbed beyond the stratum corneum and do not react with the skin (Refer to Figure 1)
If the substance comes into contact with other tissues such as leading to oral exposure or inhalation (e.g. substances applied on the face, substances that do not have requirements to wash hands after use, sprays), provide a minimum in silico analysis for mutagenicity to address lack of genotoxicity. This may not be necessary for some substances (e.g. approved food additives).
If the in silico analysis for mutagenicity is positive for the substance, then the information required will be similar to other substances that are systemically absorbed.
Microorganisms
Information to address genotoxicity is not required.
Carcinogenicity
If the proposed substance is intended to be used long-term (continuous use for at least 6 months), carcinogenicity studies are required. The toxicity profile of a substance and the indication and duration of the intended use may influence the need for carcinogenicity studies (see Guideline on repeated dose toxicity CPMP/SWP/1042/99 Rev 1 Corr).
The TGA will not generally consider an application ineffective simply because a carcinogenicity study for the substance was not provided. While in vitro mutagenicity studies have, individually, a low predictive value in terms of human carcinogenicity, any unusual results arising from a number of different in vitro and in vivo genotoxicity studies could indicate the need for further investigation. In addition, chronic toxicity studies may identify issues of concern in relation to carcinogenicity, for example, pre-neoplastic changes. For most listed medicine substances, where there is a history of human exposure through the diet or traditional medicine use, such information can assist with ascertaining its carcinogenic potential.
Exposure may occur via routes other than intended (e.g. inhalation after a sprayed dermal application); also, unusual findings in main toxicity studies may warrant further investigation. In such circumstances additional targeted toxicity studies should be considered.
Toxicity studies are normally performed for the proposed substance. However, as impurities, degradation products and metabolites may be relevant to safety assessment, specific toxicological information on these compounds may be useful if there is a safety concern related to them.
Further information about carcinogenicity studies is provided in the following scientific guidelines:
- ICH S1B(R1) – Note for Guidance on Carcinogenicity: Testing for Carcinogenicity of Pharmaceuticals CPMP/ICH/299/95
- Note for Guidance on Carcinogenic Potential CPMP/SWP/2877/00
- ICH S1C(R2) – Note for Guidance on Dose Selection for Carcinogenicity Studies of Pharmaceuticals EMEA/CHMP/ICH/383/1995
Reproductive and developmental toxicity
Data on reproductive and developmental safety is only required if there are no restrictions proposed in the application that limit use of the substance for use in pregnant or lactating females, or in a paediatric population (under 18 years).
A single, well-designed, multi-generation, prolonged-exposure reproduction and developmental study should provide sufficient information on the effects of a substance on all aspects of reproduction, including sexual behaviour, gonadal function, spermatogenic and oestrus cycles, fertility, fecundity, parturition, lactation, pre- and post-natal growth, development and maturation of the offspring. The study may also provide adequate data on teratogenesis. However, particularly if some findings in the initial multi-generation study are equivocal, separate developmental studies should be provided for embryotoxicity, teratogenicity, altered growth and the induction of functional deficits (post-natal behaviour).
For detailed information about the conduct and regulatory requirements of reproductive toxicology, applicants should refer to the relevant scientific guideline ICH S5(R3) – Detection of Toxicity to Reproduction for Medicinal Products including Toxicity to Male Fertility and OECD Guidelines for the Testing of Chemicals. For detailed information on non-clinical safety testing in juvenile animals to support paediatric indications, read Guideline on the need for Non-Clinical Testing in Juvenile Animals of Pharmaceuticals for Paediatric Indications (EMEA/CHMP/SWP/169215/2005) and ICH S11 – Nonclinical safety testing in support of development of paediatric pharmaceuticals (EMA/CHMP/ICH/616110/2018).
Presenting the data under subheadings will aid in their assessment. Typical subheadings would be, if there is information available:
- pharmacokinetics in pregnancy and lactation
- fertility and general reproductive performance
- developmental (teratology) toxicity studies
- perinatal and postnatal toxicity studies
Clinical data specifically tailored to these vulnerable populations should also be provided, if available. For detailed information, refer to ICH E11 – Note for guidance on clinical investigation of medicinal products in the paediatric population (CPMP/ICH/2711/99).
Substances for dermal use
Applications for substances that do not propose restrictions to limit use in adults only, must discuss the relevance of derivation of the MoS in children, by factoring in the difference in skin surface area over body weight ratio between children and adults. For detailed information, refer to 3-6.10 of Notes of guidance for the testing of cosmetic ingredients and their safety evaluation, 11th revision (SCCS/1628/21).
Local tolerance
Local tolerance testing should be primarily focused at the proposed sites of administration for human use. The dose, frequency and duration of exposure for the tests should closely resemble the proposed therapeutic use of the substance. In general, local tolerance testing in substances intended for oral administration is not required, provided that information to address Toxicological data have shown that the proposed substance has been in contact with the oral cavity, oesophagus and the gastrointestinal tract. The inclusion of site(s) that may come into contact with the substance through accidental exposure should also be considered. For example, a substance used in a product applied dermally to the face, normally requires assessment for eye or mucosal irritation.
See Guideline on non-clinical local tolerance testing of medicinal products (CHMP/SWP/2145/2000 Rev. 1), noting that parenteral administration is not a suitable route of administration for listed medicine substances.
Substances for dermal use (refer to Figure 1)
The following studies are required:
- Skin irritation study: OECD 404 (acute dermal irritation), OECD 439 (in vitro skin irritation) or human repeat patch insult test (HRPIT)
- Skin sensitisation study: OECD 406 or HRPIT
- Eye irritation study: OECD 405 (acute eye irritation/corrosion) or OECD 492 (reconstructed human cornea-like epithelium).
- Photosafety studies (phototoxicity and photosensitisation) if intended for dermal use where there is potential for sun exposure. For detailed information, refer to 3-4.12 of Notes of guidance for the testing of cosmetic ingredients and their safety evaluation, 11th revision (SCCS/1628/21).
- Phototoxicity is not required if the Molecular Extinction Coefficient (MEC) of the substance is below 1000 L mol-1 cm-1.
- In vitro phototoxicity testing is not required if the substance only absorbs at wavelengths lower than 313 nm and if there is insufficient absorption at longer wavelengths.
- Photomutagenicity tests may be requested if the phototoxicity tests are positive.
If local tolerance studies have not been undertaken in broken skin or in the eye, respective restrictions ‘not to be included in medicines intended for use on broken skin’ (or words to that effect) or ‘not to be included in medicines intended for use in the eye’ (or words to that effect) will apply to the proposed substance.
Adverse reactions
Submit all reports, published and unpublished relevant to the safety of the proposed substance. Include information on the nature, severity and frequency of adverse reactions and information on potential interactions of the substance with food or medicines. Reports of poisonings (e.g. accidental poisoning or suicide attempts) must be provided with details of doses consumed, the specific form of the substance (e.g. sodium selenate and selenomethionine) and the circumstances of the poisoning (e.g. inadequate closures on bottles or chronic toxicity via the diet). It is acknowledged that clinical studies of longer duration and those involving large numbers of subjects may result in adverse events. Whether the adverse reactions differ between intervention groups will determine if such events are treatment related.
When searching for reports of adverse reactions, use known synonyms for the substance and, if relevant, for closely related substances or components of the substance e.g. for kava, the search should include, among other things, Piper methysticum, kava, Piper inebrians, kavain, dihydrokavain and methysticin.
The TGA’s Database of Adverse Event Notifications and Health Canada’s Canada Vigilance Adverse Reaction Online Database can be searched by the ingredient or brand name of drugs and health products. Similarly, WHO’s VigiAccess™ also has a database which can be useful for information on medicinal products. Rapex - Rapid Alert System for dangerous nonfood products is another system which collects adverse outcome data on cosmetics marketed in Europe and can be searched for products known to contain the substance that is the subject of the application. Likewise, the FDA has a CFSAN Adverse Event Reporting System which contains information on adverse event and product complaint reports for foods, dietary supplements and cosmetics.
Searches should be performed for adverse reactions in all countries where the substance is in use. Adverse reaction reports obtained from national medicine safety surveillance authorities should include a description of all available clinical information and the outcome of the reaction. From these reports, an applicant can identify products that contain the substance and consider the relevance of any reports to the substance. If there are several such reports, the narratives should be included as an attachment. Provide a summary table as per Table 23.
Note, the absence of adverse events in products alone is not sufficient evidence to substantiate the safety of a new substance, however they can be used as supporting information. Particularly for overseas regulated products, there are numerous other factors that must be considered, such as:
- Mandatory reporting in those markets
- Types of products (dosage, dose form, population. indications etc.) the substance was used in
- Influence the formulations have on the occurrence or lack of adverse events reported
- Existence of labelling directions and controls on those products
- Number of units sold in that market
- The demographic and potential number of consumers who are likely to have used those products, and duration of availability in that marketplace.
Table 23: Example format – Summary of adverse events
| Report reference and date reported | Patient details | Product details | Treatment details | Other meds | Adverse reaction | Comments e.g. outcome; lab results | Causality |
|---|---|---|---|---|---|---|---|
| Adverse Drug Reactions Systems (Aust) Report No. 24369 30-6-98 | Male 34 years | Brand name Ingredients: (active ingredient details) | 480 mg tablets PO 3 times daily for 10 weeks | Aspirin off and on; cod liver oil 275 mg PO twice daily | Psychosis (psychotic ideation); manic reaction (hypomania) | Recovery after (Brand name) stopped | Probable |
| BfArM 9204235 (Germany) 16-6-92 | Female 59 years | Brand name Ingredients: (active ingredient details) | 200 mg capsules twice daily PO | Serepax 45 mg daily PO | Headache; impaired alertness; amnesia, nausea | Recovery after all medication stopped | Possible |
Acronyms used: PO: (per os) oral administration
Substances of human or animal origin
Substances that are of human or animal origin, or that are used during manufacturing, with potential viral and Transmissible Spongiform Encephalopathy (TSE) risks must provide a clearance of risk from TSE.
For further information refer to Transmissible Spongiform Encephalopathies (TSE): TGA approach to minimising the risk of exposure.
Microorganisms
Information for this section is not required.
Appendix
Table 34: Summary of core information requirement for different substance types
- This table is applicable to new substances for use in listed medicines (including complementary medicines, sunscreens, topical medicines and oral health products) not subject to a monograph in a default standard.
- Careful consideration has been given to dermal excipients and the following core information are considered critical for reasons attached:
- Identity defines the substance that has been assessed
- Assay establishes the quantity of the substance that has been assessed to be safe
- Impurities and incidental constituents testing is critical to inform the ingredient-related impurities that are not identifiable in a finished product
- The Compositional guideline is important where there is no corresponding monograph in a default standard, for the purposes of Good Manufacturing Practice and safety; it provides clarity of the nature and quality of the ingredient that has been evaluated and approved to be safe.
| Core information requirement | Substances for oral use3 | Microorganisms4 | Dermal active substances5 | Dermal excipient substances5 | ||
|---|---|---|---|---|---|---|
| QUALITY | ||||||
| Description | Description of the substance | Required | Required | Required | Required | |
| Manufacturing details | Description of manufacturing process | Required | Required | Required | Required6 | |
| Control of materials | Required | Required | Required | Not required | ||
| Critical steps & intermediates | Required | Required | Required | Not required | ||
| Process development | Required | Required | Required | Not required | ||
| Process validation | Required | Required | Required | Not required | ||
| Characterisation | General properties | Required | Required7 | Required | Required | |
| Identity | Required | Required | Required | Required | ||
| Assay | Required | Required | Required | Situational8 | ||
| Impurities and incidental constituents | Required | Required | Required | Required | ||
| Reference standard | Required | Required | Required | Situational9 | ||
| Specifications | Compositional guideline | Required | Required | Required | Required | |
| Certificates of Analysis (CoA) | Active | Active | 2 commercial-scale OR | 1 commercial-scale OR | ||
Excipient | Excipient |
|
| |||
| Stability test | Real-time and accelerated stability testing data10 for two commercial-scale batches or three pilot scale batches | Required | Required | Required | Not required | |
| Core information requirement | Substances for oral use3 | Microorganisms4 | Dermal active substances5 | Dermal excipient substances5 | ||
| SAFETY | ||||||
| Systematic literature search | A systematic literature search on the substance; with the search strategy and results with justification for inclusion / exclusion of data | Required | Required | Required | Required | |
| History and pattern of human use | Information on:
| Required | Required | Required | Required | |
| Biological activity | Pharmacokinetics | Pharmacokinetic studies addressing:
| Required | Situational11 | Required | Required |
| Required | Situational11 | Situational12 | Situational12 | ||
| Required | Situational11 | Situational12 | Situational12 | ||
| Required | Situational11 | Situational12 | Situational12 | ||
| Pharmacodynamics | For substances that are systemically absorbed (or cannot be excluded), pharmacology information addressing:
| Required | Situational13 | Situational14 | Situational14 | |
| Required | Situational13 | Situational14 | Situational14 | ||
| Required | Required15 | Situational14 | Situational14 | ||
| Core information requirement | Substances for oral use3 | Microorganisms4 | Dermal active substances5 | Dermal excipient substances5 | ||
| Toxicological data | Information from in vitro studies, animal studies, human clinical studies or other information (or a combination) addressing:
| Required | Required | Situational16 | Situational16 | |
| Required | Required | Situational16 | Situational16 | ||
| Required | Not required | Situational16 | Situational16 | ||
| Required | Required | Situational16 | Situational16 | ||
| Required | Required | Situational16 | Situational16 | ||
| Required | Required | Required | Required | ||
| Not required | Not required | Situational17 | Situational17 | ||
| Adverse reactions | A list of the nature, severity and frequency of adverse reactions from adverse event databases, clinical trials or case reports of human poisoning | Required | Required | Required | Required | |
| Substances of human or animal origin | Information on clearance of risk for transmissible spongiform encephalopathy (TSE) if substances of human or animal origin were used during manufacture | Required | Not required | Required | Required | |
Footnotes
- ICH Q3D is referenced in the BP general notice <2034> ‘Substances for Pharmaceutical Use’ for permitted daily exposures (PDE) for elemental impurities.
- Permitted Daily Exposure
- Substances which are generally for oral use and any other forms not specified in the table.
- Microorganisms are whole and intact cells of bacteria and fungi (including yeasts) that are live or non-viable.
- Includes substances for use in listed sunscreens and topical listed medicines.
- For chemically derived excipients for dermal use only, only a brief description of manufacturing process required.
- For live microorganisms, information addressing antimicrobial resistance and susceptibility, information to address absence of virulence factors, toxigenic and pathogenic attributes is also required.
- An assay test is not a requirement if the excipient is only for use in a formulation for its physical properties.
- A reference standard is not a requirement for chemically derived excipients for dermal use only.
- Stability testing is not required if stress testing data can be provided that demonstrates the absence of degradants.
- Only information to demonstrate absence of microorganism in the systemic circulation in the host is required.
- This information is not required if the substance is for use on unbroken skin only, and is demonstrated to not be absorbed beyond the stratum corneum.
- Only information addressing mechanisms of action is required.
- This information is not required if the substance is for use on unbroken skin only, is demonstrated to not be absorbed beyond the stratum corneum, and does not react with the skin.
- For live microorganisms, only information addressing pharmacodynamic interactions with antibiotics/antifungals and host immunity is required. For non-viable microorganisms, only information addressing pharmacodynamic interactions with host immunity is required.
- This information is not required if the substance is for use on unbroken skin only, is demonstrated to not be absorbed beyond the stratum corneum, and does not react with the skin.
- This information is only required if genotoxicity data has not been provided
Print version
Page history
Title changed from 'Application requirements for new substances in listed medicines' to 'Understanding the application requirements for a new substance in listed medicines' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Original publication, authored by the Complementary and Over the Counter Medicines Branch.
- Merging of ‘Applications for new substances in listed medicines’ and ‘Information required in an evaluation of a substance for use in listed medicines: guidance for sponsors’ to consolidate information for applicants. All content reviewed and updated for correctness, to reduce duplication and improve clarity.
- Amendments to previous guidance to reflect the new documents ‘Mandatory requirements for an application to vary the Permissible Ingredients Determination’, ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’ and ‘Guidance on using evaluation reports from Comparable Overseas Bodies’.
- Inclusion of subheadings with tailored guidance for different substance types.
- Inclusion of an Appendix summary table for core information required for different substance types.
Title changed from 'Application requirements for new substances in listed medicines' to 'Understanding the application requirements for a new substance in listed medicines' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Original publication, authored by the Complementary and Over the Counter Medicines Branch.
- Merging of ‘Applications for new substances in listed medicines’ and ‘Information required in an evaluation of a substance for use in listed medicines: guidance for sponsors’ to consolidate information for applicants. All content reviewed and updated for correctness, to reduce duplication and improve clarity.
- Amendments to previous guidance to reflect the new documents ‘Mandatory requirements for an application to vary the Permissible Ingredients Determination’, ‘Requirements for microorganism characterisation in listed medicines and registered complementary medicines’ and ‘Guidance on using evaluation reports from Comparable Overseas Bodies’.
- Inclusion of subheadings with tailored guidance for different substance types.
- Inclusion of an Appendix summary table for core information required for different substance types.