Applying for a Conformity Assessment certificate for medical devices
An overview on the application process for manufacturers aiming to obtain a TGA-issued Conformity Assessment certificate.
Purpose
This guidance document provides an overview on the application process for manufacturers aiming to obtain a Conformity Assessment certificate issued by the Therapeutic Goods Administration (TGA).
It covers key aspects such as how to navigate the process effectively, application timeframes, the role of case management, component assessment against the Essential Principles, and includes details regarding the TGA quality management system (QMS) assessment.
TGA Conformity Assessment applications
Manufacturers make TGA Conformity Assessment applications for new certification, substantial changes to an existing certificate, and recertification of a certificate that is about to expire.
Guidance on how to make an application for TGA Conformity Assessment certification, is available at Application instructions: Conformity Assessment certification.
Processing times
All Conformity Assessment applications, including those for in-vitro diagnostic devices, are required to be processed within 255 working days as specified in Regulation 4.3 of the Therapeutic Goods (Medical Devices) Regulations 2002 (the Regulations).
We use processing times to help us plan our work, this is beneficial for both the TGA and Industry. Applications are evaluated as soon as possible, with priority given to those applications with longer processing times and those containing all necessary information.
Recent processing times for Conformity Assessment applications are published in our Annual Report and on the TGA website.
Target timeframes for Conformity Assessment applications
Through our consultations with Industry, we now have target timeframes for applications, we aim to meet these targets in 90% of applications.
| Application type | Target timeframe business days with TGA | Maximum timeframe business days with TGA | Applicant dates business days with applicant (contingent on number of questions raised per round) | Estimated total timeframe (includes total applicant days) |
|---|---|---|---|---|
| New or substantial | 160 business days | 200 business days | 40-100 business days | 10-15 months total (~300-445 days) |
| New or change applications with ACMS* advice OR requiring on-site TGA audit of facilities | 190 business days | 225 business days | 40-100 business days | 12-16 months total (~330-480 days) |
| Recertification | 80 business days | 150 business days | 40-60 business days | 6-10 months total (~185-310 days) |
* Advisory Committee on Medical Devices (ACMD).
To meet these timeframes, we have implemented the following processes:
- engagement with the applicant throughout the process with a dedicated case manager
- improved resourcing and internal processes to conduct component assessments concurrently
- requests for additional information are synchronised so that you receive a request that incorporates multiple component areas (clinical, engineering etc) where necessary
- limiting the number of additional requests for information for recertification applications. We’ve modified the supporting data form, with clear guidance to manufacturers on the information we need to process these applications
- limited the use of mutual stop of the application clock (see below)
- we will not accept changes to the scope of the application after the pre-assessment stage.
Application clock
We monitor the processing time for each application using an “application clock” that:
- Starts once the application has been submitted via the TGA Business Services Portal and the Application fee has been paid. This is also referenced as an ‘effective application’, and the clock starts counting from day zero.
- Operates during business days, excluding ACT public holidays and weekends.
- Stops on specific occasions as outlined below.
- Applicants can reach out to their case manager who can provide you with a status update for the application.
- The application clock is stopped in the following circumstances:
- When we request additional information necessary for evaluation, such as through a request under section 41JA of the Therapeutic Goods Act 1989 (the Act) or under Regulation 4.3(3)(a). The clock restarts when we receive all the required information.
- When we invoice for the assessment fee - the clock restarts when the fee is paid.
The application clock may also be stopped in mutually agreed extenuating circumstances, such as:
- manufacturing facility affected by a natural disaster like severe flood, earthquake, or fire
- death or serious illness of critical personnel responsible for obtaining information.
Extenuating circumstances does not include absence due to holidays, missed requests from the TGA, or time to translate documents or generate missing information.
Conformity Assessment process flow
The Conformity Assessment application process is outlined in detail below and shown in Figure 1.
Pre-submission meeting
To support applications being processed as fast as possible, we encourage all applicants to engage with the TGA early.
This is especially important for new manufacturers and those with novel or emerging technology.
The benefits of a pre-submission meeting for the applicant and the TGA are:
- a common understanding of the medical device
- potential insights for new or novel device
- clarification of the appropriate application type and pathway
- clear understanding of required supporting documents
- early opportunity to receive critical feedback
- resolution of issues before submitting application
- opportunity to map and manage timeframes and required resources.
Submission
The applicant (the manufacturer, their agent, the sponsor or representative) submits a Conformity Assessment application via the TGA Business Services portal.
Applicants are required to provide evidence in the form of a technical file or design dossier, depending on the medical device classification, and pay the application fee.
Following the applicant’s submission of their Conformity Assessment application via the TGA Business Services Portal and paid application fee.
We will send the applicant a request for the supporting information.
This request may require you to complete a Supporting Data Form – Conformity Assessment Certification and provide the associated data.
The form and associated data should only be provided when requested.
The appropriate supporting data forms are available under ‘Forms’ in the Conformity Assessment application.
Pre-assessment
Following payment of the application fee and once all supporting data has been received, the application proceeds to pre-assessment.
We aim to complete pre-assessment within 30 business days.
During pre-assessment, assessors verify information such as:
- whether your product is a medical device
- whether your device has been correctly classified
- whether the Conformity Assessment procedure applied is suitable for the class of the device
- whether sufficient information submitted to enable pre-assessment
- the number of UPIs and models in scope of assessment.
An assessment plan is developed at the pre-assessment stage and outlines the types of specialist component assessment required.
Your application may also be screened at this stage by the specialist component areas to identify any major gaps or missing critical information.
Following screening you will receive a feedback email from the TGA which outlines:
- what component assessment areas will be required for your application
- if an assessment of the manufacturer’s quality management system is required and whether that will be an on-site audit or off-site (desktop) assessment
- whether we are likely to require expert advice from the Advisory Committee on Medical Devices (ACMD) for your device
- any gaps identified by the assessor during screening. If we identify gaps during the screening, we will send you a formal request to provide the missing information.
The assessment plan and the scope of the application determines the assessment fees, which we will invoice after we complete the pre-assessment.
When you pay the fees, we will assign a case manager to your application and start each of our component assessments.
Round 1
The case manager coordinates the assessments required for your application with the relevant component areas.
Our aim is for all component assessment areas to complete a round 1 evaluation within 100 business days.
Each component area will advise the case manager about any deficiencies in the data and the case manager will then send you a request for information, if required. Your case manager will set you a timeframe to respond (between 20 – 40 business days) depending on the volume and complexity of the questions.
If you wish to seek clarification of the questions raised or discuss the progress of the application, you can contact the case manager or request a meeting.
The case manager may also contact you for further discussion if this would help progress the application.
If your application contains all sufficient information following the first round of assessment, the application will then proceed for decision by the delegate.
Round 2
After we receive your response to the round 1 request for information, the case manager will forward your responses to the relevant component assessment areas.
If there is insufficient data provided in the response, you may receive additional questions to clarify your response.
Again, your case manager will forward you the consolidated list of questions for you to respond in 20- 40 business days, based on the complexity of the questions.
Throughout the process you will be aware of any issues raised and will have the opportunity to discuss these.
The formal request for information provides you the opportunity to submit information for consideration by the delegate.
Advisory Committee on Medical Devices
Some Conformity Assessment applications for high-risk devices, or devices with novel technology, may involve consultation with the Advisory Committee on Medical Devices (ACMD).
The ACMD is a panel of academic and clinical specialist experts and includes a consumer representative.
If we seek advice from the ACMD, the applicant will be advised prior to the meeting.
Following the meeting, the applicant will be provided the questions and the responses provided by the committee.
We may seek advice from the ACMD at any stage of the application process outlined above prior to a decision.
Decision
Case managers will review the evaluations from all component assessment areas. They will present the outcomes to the delegate to support them in making a decision.
The delegate considers all the information related to the application and makes a decision on whether to issue a Conformity Assessment certificate.
If the delegate refuses to grant you a Conformity Assessment certificate, they will include a statement of reasons for the decision.
If a Conformity Assessment certificate is not granted there is no provision for a refund of the application or assessment fees.
Notification
We will inform the applicant of the outcome, and the application will be closed.
At any stage during assessment of the application we may seek external advice from the ACMD or other experts. We may also refer to external evaluators for advice for novel technologies, if required.
Case management
Once the application fee is paid, the application will be assigned to a case manager, who will then reach out to the applicant and introduce themselves as being the primary contact for the duration of that application process.
Each application will have a case manager as a point of contact throughout the assessment process.
If the assigned case manager leaves the TGA or takes extended leave, the applicant will be notified and provided a new case manager.
Case managers will engage with the applicant by requesting further information to address any deficiencies raised by the individual component assessment areas.
Case managers will follow up with the component assessment areas to ensure they are meeting target timeframes.
If a component area requests advice from the ACMD, they will advise the case manager. Case managers will:
- communicate with the applicant
- facilitate meetings where required
- liaise with quality management system (QMS) auditors
- advise the applicant if the application will go to the ACMD for advice
- consolidate requests for information from the component assessment areas
- track and monitor progress of the application
- conduct certain aspects of assessment, such as checking compliance with labelling requirements.
Figure 1. Conformity Assessment application evaluation process
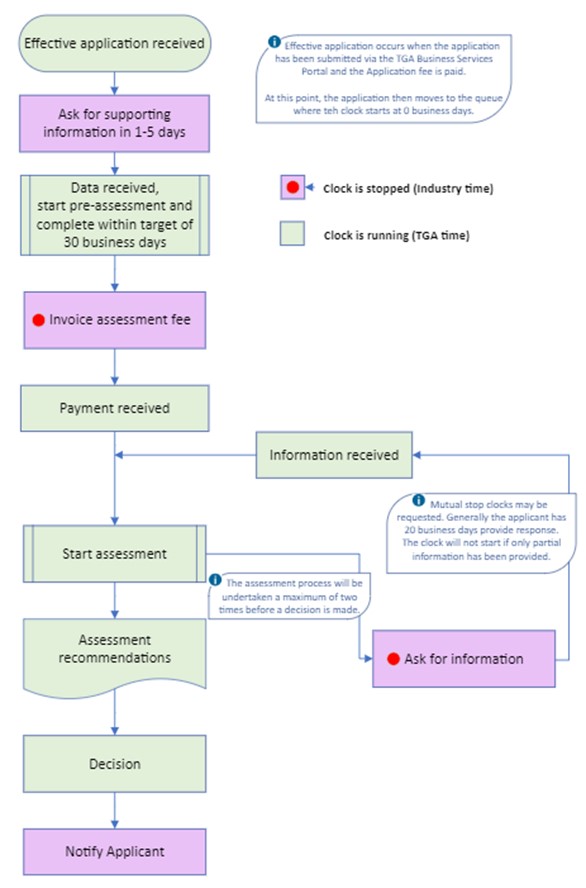
{kind=link}
Flow chart depicting the Conformity Assessment application evaluation process:
- Start: Effective application received
- Note: Effective application occurs when the application is submitted via the TGA Business Services portal and fee paid. At this point, the application then moves to the queue where the clock starts at 0 business days.
- Ask for supporting information in 1-5 days.
- Clock is stopped (Industry time)
- Data received, start pre-assessment and complete within target of 30 business days.
- Clock is running (TGA time)
- Invoice assessment fee
- Clock is stopped (Industry time)
- Payment received.
- Information received.
- Note: Mutual stop clocks may be requested. Generally the applicant has 20 business days to respond. The clock will not start if only partial information has been provided.
- Start assessment.
- Note: The assessment process will be undertaken a maximum of two times before a decision is made.
- Assessment recommendations.
- Ask for information.
- Loops back to 'Information received
- Decision
End: Notify Applicant.
The process flows linearly from top to bottom, with one loop from 'Ask for information' back to 'Information received'. Steps where the clock is stopped (Industry time) are highlighted in purple, while steps where the clock is running (TGA time) are in light green.
Quality management system (QMS) audits
As part of the Conformity Assessment process, we audit the manufacturer’s QMS for manufacturing the kinds of medical devices in the application.
We audit against regulatory requirements and internationally recognised standards, such as ISO13485:2016.
We may conduct on-site audits, at the premises relevant to the manufacture of the medical device, or off-site audits of documents provided with the application, known as desktop assessments.
We will tell you the type of QMS assessment we will need to do, at the end of the pre-assessment stage when we invoice the assessment fees.
We will normally do an on-site QMS audit at one or more facilities, depending on the specific details of the manufacturing processes.
We may abridge the assessment and do a desktop assessment in some cases as described below. This section provides an overview of our QMS audit processes.
For more information, see Quality management system audits and certification guidance.
On-site audit
On-site QMS audits will be conducted at the premises identified as relevant to the manufacture of the medical devices in the application.
The auditor will visit the facility and observe the processes performed there. Auditors will also review documents related to processes and procedures and conduct interviews to verify the processes and procedures have been applied.
At the conclusion of the audit, the auditor will present the manufacturer with initial findings. The auditor will provide an audit report within 60 days after the audit, detailing any nonconformities identified.
The manufacturer must address the nonconformities with corrective actions and provide the auditor with evidence that the nonconformities have been addressed.
We also conduct surveillance audits after the Conformity Assessment certificate has been issued.
On-site surveillance audits assess continual compliance with the requirements and the conditions imposed by the Conformity Assessment certificate.
A manufacturer issued with a TGA Conformity Assessment certificate will be subject to surveillance audits by the TGA.
Off-site desktop assessment
The TGA is a participating member of the Medical Device Single Audit Program (MDSAP) and considers audit results from other comparable overseas regulators such as an EU notified body.
If the application includes evidence that the manufacturer has been audited and certified by a comparable overseas regulator, such as audit reports and close-out records, we will evaluate that evidence to ensure that the audit activities were aligned with Australian regulatory requirements and that the audit activities assessed the manufacturing processes and kinds of medical devices in the Conformity Assessment application.
Further assessment, including an on-site audit, of a manufacturer’s QMS may be undertaken if compliance is not able to be determined.
We also assess the manufacturer’s quality manual, procedures, records, and reports (e.g., process validation, risk management, post-market surveillance) that were provided with the application.
We normally assess substantial changes to a manufacturer’s QMS via a desktop assessment.
An off-site QMS desktop assessment could result in a request for further information and a second round of the assessment.
The desktop assessment could recommend that we do an on-site QMS assessment if we have insufficient evidence that the QMS requirements have been satisfied.
Component assessments
The case manager will actively oversee the component assessments outlined during pre-assessment. Technical experts from each area will assess the information provided about the medical device to ensure it complies with the EPs.
Advice and other assessments may be sought from additional areas of the TGA depending on the device characteristics.
Below is a general summary of what the main component area looks at in more detail.
Biomaterials
The biomaterials component evaluates the relevant supporting scientific evidence and data provided for biomaterial and material manufacture and safety against EPs 2, 5, 7, 9, 13 and 13A.
The assessment reviews the material composition, design, and manufacture (including sterilisation processes, packaging materials, and stability).
The assessment focuses on the device’s interaction with the body and the duration of use.
Engineering
The engineering component evaluates the evidence related to the physical and mechanical properties of a medical device against EPs 1-5, 7.1(a), 9.1, 9.2, 12, 13, 13A and 13B.
The evaluation may cover the following device safety and performance aspects considering the acknowledged state of the art for the device:
- risk management
- post-market data
- device verification
- validation test reports (bench tests, shelf-life, packaging integrity, usability, particulates)
- labelling, implant patient information materials, and instructions for use.
Software
A software assessment is undertaken on medical devices incorporating software or software as a medical device. The assessment will focus on compliance with EPs 12.1, 13.2 and 13B specific to software and include assessment of:
- artifacts for design and architecture
- verification and validation of software
- defect management process
- human factors and usability
- cybersecurity and privacy
- interoperability of devices.
In addition to general software requirements, for software that uses artificial intelligence (AI) or machine learning (ML) algorithms, the manufacturer is required to show evidence that is sufficiently transparent to enable evaluation of safety and efficacy of the product.
Transparency means that a ‘black box’ approach would not be considered acceptable (i.e., the TGA will not accept that no evidence can be provided because it is ‘black box’ technology).
- While this is a rapidly evolving area, this will typically include artefacts for the following: Overarching statement of the objectives of the AI/ML model.
- Algorithm and model design, including techniques used.
- Data used for training and testing machine learning models – and generalisability where applicable.
- Synthetic data would not be considered suitable for all clinical use cases.
- Size of data sets must be sufficiently large to be statistically credible.
- Information about populations that this data is based on and justification for how this data would be appropriate for the Australian population who will be using the device.
Additional guidance is available on the regulation of software-based medical devices and software-based medical devices FAQs.
Clinical
The clinical component assesses the clinical evidence for the medical device, considering EPs 1, 2, 3, 4, 6, 13, 13A and 14.
The level of clinical evidence should be proportionate to the device classification, type, and intended purpose.
The assessment will check that the clinical evidence demonstrates that the device functions as intended and has an acceptable benefit-risk profile. The clinical evidence must demonstrate the device performance aligns with its intended use. We consider the current scientific understanding related to the device treatment approach.
The clinical evidence must show that all potential risks are minimised during design and development, with any remaining risks outweighed by the benefits.
Additionally, product labelling should reflect the clinical evidence, and risk management documents must detail how residual risks are mitigated.
Our clinical evidence guidelines outline:
- what constitutes clinical evidence
- the process of clinical data generation
- clinical evaluation to produce the clinical evidence.
In vitro diagnostics (IVD)
The IVD component evaluates the quality, safety and performance of IVD medical devices, assessing against EPs 1-6, 8, 9, and 12-15.
We focus on:
- intended purpose
- stability
- labelling
- risk management
- clinical performance
- analytical performance.
Our assessment will vary depending on the intended purpose of the device (diagnosis, screening, or monitoring) and on the intended user of the device (laboratory, health professional, or lay person).
Microbiology and sterility
The microbiology component includes assessment of the manufacturing and sterilisation details. We primarily assess compliance with EP 8, which covers sterile or non-sterile medical devices.
Our evaluation of the scientific evidence focuses on compliance to internationally recognised standards and best scientific practices.
For instance, we may reference ISO 11135 for ethylene oxide, the ISO 11137 series for radiation processing, and AAMI TIR 56 guidance for ethylene oxide in a flexible chamber.
Page history
Title changed from 'Conformity assessment certificate process and timeframes' to 'Applying for a Conformity assessment certificate for medical devices' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Original publication.
Title changed from 'Conformity assessment certificate process and timeframes' to 'Applying for a Conformity assessment certificate for medical devices' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Original publication.