Transitioning to new manufacturer evidence for in-vitro diagnostic medical devices (IVDs)
This guidance is to help sponsors and manufacturers of in-vitro diagnostic (IVD) medical devices transition to new manufacturer evidence requirements.
Purpose
This guidance is to help sponsors and manufacturers transition to the new manufacturer evidence requirements for in vitro diagnostic (IVD) medical devices.
Manufacturer evidence are documents, usually certificates or records of regulatory approval by a comparable overseas regulator, that are submitted to the Therapeutic Goods Administration (TGA) to support medical device entries in the Australian Register of Therapeutic Goods (ARTG).
Sponsors will need new manufacturer evidence for their IVD medical devices because of:
- the new European Union IVD Regulation 2017/746 (EU IVDR) that replaces the IVD Directive 98/79/EC (IVDD)
- the end of the transition period for the TGA to accept ISO 13485 certificates as manufacturer evidence, unless supported by the manufacturer’s EU Declaration of Conformity made under the IVDD, before 26 May 2022.
Sponsors of existing IVD ARTG entries that need to transition to new manufacturer evidence will need to act to ensure ongoing regulatory compliance. The extent of those actions will depend on a range of factors such as whether the devices covered under the new certification, or the intended purpose of those devices, change.
This guidance covers a range of scenarios and examples to help identify the correct pathways and the sponsor actions required.
Sponsors may need to submit a variation application, and in rare cases, recall action may also need to be considered.
Legislation
Background
The TGA accepts a range of manufacturer evidence to support inclusion of IVD medical devices in the ARTG, including certification and approvals from EU and other comparable overseas regulators.
Information on the range of manufacturer evidence that can be used to support an ARTG inclusion can be found in TGA legislation and guidance.
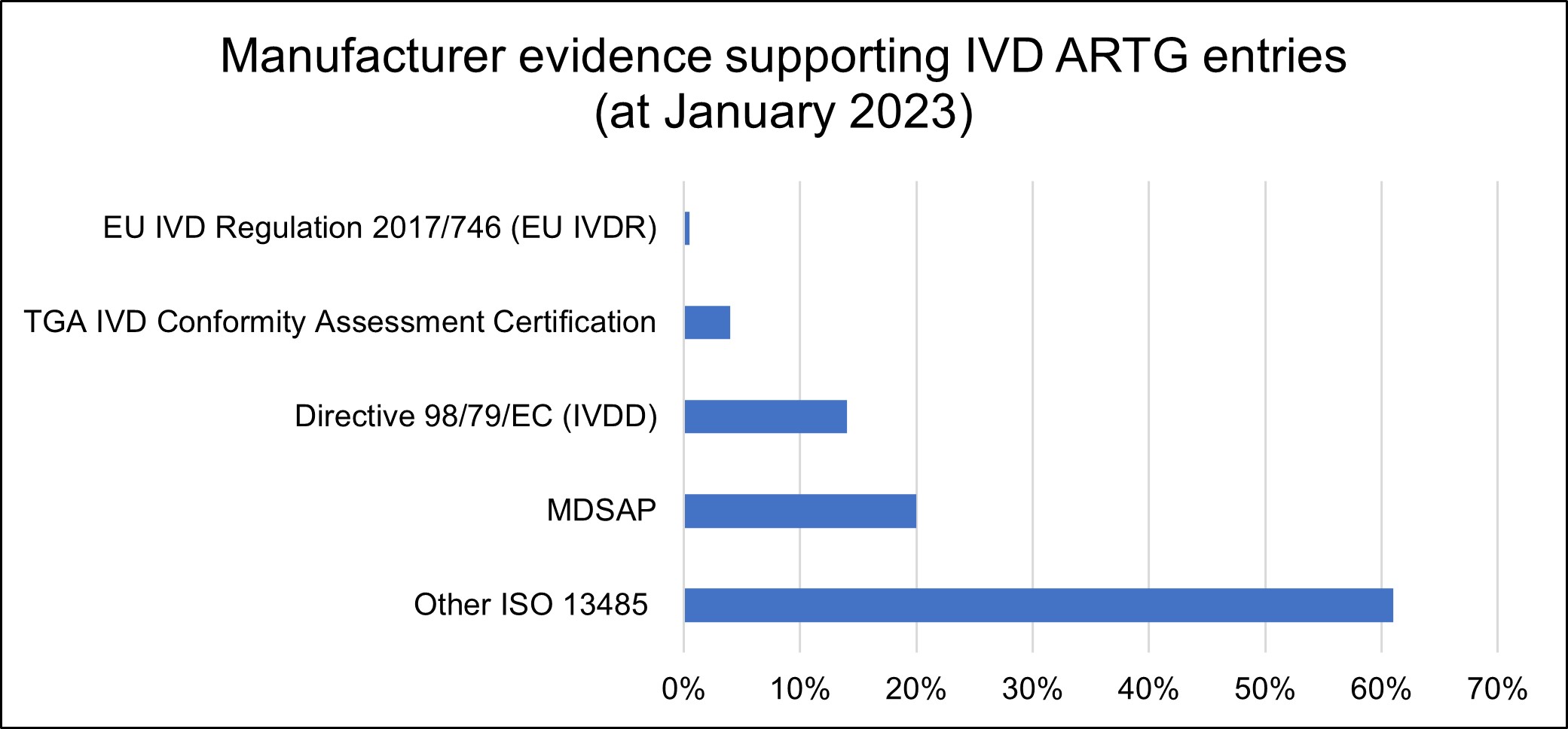
A breakdown of the types of manufacturer evidence submitted to the TGA is illustrated in the graph below:
Manufacturer evidence graph
{kind=link}
Bar chart showing manufacturer evidence supporting IVD ARTG entries as of January 2023.
The graph shows five categories with their corresponding percentages:
- EU IVD Regulation 2017/746 (EU IVDR): approximately 2%
- TGA IVD Conformity Assessment Certification: approximately 5%
- Directive 98/79/EC (IVDD): approximately 15%
- MDSAP: approximately 20%
- Other ISO 13485: approximately 60%
The bars are displayed horizontally in blue, with percentages shown on the x-axis from 0% to 70%."
The changes described in this guidance will impact about 70% of IVD ARTG entries, particularly those supported by ISO 13485 certificates and certification issued under EU IVDD. Over time, we expect those forms of certification to be replaced mostly by certification issued under EU IVDR, Medical Device Single Audit Program (MDSAP) certification or any other comparable overseas regulator certification accepted by the TGA.
This guidance describes transitions in the types of certificates that the TGA can accept as manufacturer evidence and does not indicate a change in the status of the international quality management system standard for medical device regulatory purposes: ISO 13485. The TGA continues to recognise ISO 13485, as a standard, as before.
However, as detailed in this guidance, the TGA no longer accepts ISO 13485 certificates issued under the International Accreditation Forum framework.
Changes to manufacturer evidence requirements
Transition from EU IVDD to EU IVDR
The EU IVD Medical Devices Directive (EU IVDD) is being replaced by the new EU IVD Regulation (EU IVDR). This means sponsors of IVD medical devices included in the ARTG based on certification issued under EU IVDD will need to transition those entries to certification issued under EU IVDR or another acceptable certification (for example, MDSAP - Medical Device Single Audit Program certification) to continue to supply those IVDs in Australia. The EU IVDR was published in May 2017 and on 24 April 2024 the EU extended the transition period, subject to eligibility conditions, until:
- 31 December 2027 for class D devices
- 31 December 2028 for class C devices
- 31 December 2029 for class B and class A sterile devices.
The EU IVDR introduces a range of changes for IVD medical device manufacturers such as:
- more stringent requirements to demonstrate IVD medical device safety for patients and users including requirements for performance and clinical evidence
- additional requirements for the manufacturer’s quality management system
- changes to labelling and instructions for use requirements
- potential changes to the scope of indications or intended purpose which could change the classification of the IVD medical device.
We will continue to accept EU IVDD evidence to support new IVD applications for inclusion in line with the EU transition periods when the sponsor provides evidence that the manufacturer meets the EU IVDR extension eligibility conditions. A summary of relevant cut-off dates is included in the table below (Table 1).
A valid certificate refers to a certificate that is within expiry and covers the scope of the IVD devices included in the application.
If the sponsor is not able to obtain the conformity assessment evidence within the transition timelines, refer to expiring or lapsing manufacturer evidence section of this guidance for actions required.
Note
Sponsors and manufacturers do not need to transition to certification under EU IVDR, if their ARTG entries and IVD medical devices are currently supported by other valid certification (e.g. MDSAP) accepted by the TGA for that class of device.
Transition from ISO 13485 with an EU Declaration of Conformity
Except for certain higher risk devices, most IVD medical devices were self-declared under the EU IVDD and did not require certification for supply within Europe.
However, to supply these IVD medical devices in Australia, manufacturers needed to obtain certification, such as ISO 13485 certification from an EU notified body or a third-party accredited conformity assessment body. We required the sponsor to provide evidence of this certification and the manufacturer’s EU Declaration of Conformity made under the IVDD for devices supplied in Australia.
ISO 13485 certificates result from quality management system assessments and are not associated with a regulatory review of the manufacturer’s IVD medical devices. For this reason, the TGA is phasing out acceptance of ISO 13485 certificates on its own in favour of other forms of evidence.
In line with the implementation dates of the EU IVDR in Europe, the TGA will stop accepting ISO 13485 certificates supported by an EU IVDD Declaration of Conformity from:
- 26 May 2026 for class 3 IVDs
- 26 May 2027 for class 2 IVDs.
These transition periods only apply where the manufacturer’s EU Declaration of Conformity was made under the IVDD before 26 May 2022, and covers the devices included in the application to the TGA.
Transition from ISO 13485 certification
We will continue to accept ISO 13485 certificates associated with a Declaration of Conformity issued under EU IVDD, since those devices will have EU regulatory scrutiny, during the IVDR transition.
However, in all other cases, ISO 13485 certificates will not be accepted to support new applications for class 2 and 3 IVD medical devices.
Existing ARTG entries supported by a valid ISO 13485 certificate can continue using that certificate as evidence until it expires. We strongly encourage sponsors to obtain another form of valid manufacturer evidence to support those ARTG entries before the associated ISO certificate expires.
Note
MDSAP certificates that cover ISO 13485 requirements are accepted.ISO 13485 certificates issued by a body accredited by a signatory to the Multilateral Recognition Arrangement of the International Accreditation Forum, are no longer accepted for new IVD inclusion applications.
ISO 13485 certificates issued by a Notified Body designated under the EU IVDD and supported by the manufacturer’s EU Declaration of Conformity made under the IVDD before 26 May 2022 are accepted.
Summary of TGA cut-off dates
The TGA cut-off dates for accepting different types of manufacturer evidence to support IVD medical device applications are summarised below.
Manufacturer evidence (QMS certification and evidence of product review) | TGA cut-off dates |
|---|---|
| ISO 13485 certificate | Not accepted after 26 May 2023 |
Certificate issued under EU IVDD The sponsor may also need to provide evidence that the manufacturer meets the EU IVDR extension eligibility conditions.
| Class 4 IVD until 31 December 2027 Class 3 IVD until 31 December 2028 Class 2 IVD until 31 December 2029 |
| ISO 13485 certificate with EU Declaration of Conformity (made before 26 May 2022) under EU IVDD | Class 3 IVD until 26 May 2026 Class 2 IVD until 26 May 2027 |
| TGA conformity assessment certificate | Ongoing acceptance |
| Certificate issued under EU IVDR | Ongoing acceptance |
| MDSAP certificate (for Class 2 and 3 IVDs) | Ongoing acceptance |
MDSAP certificate with acceptable product review evidence:
| Ongoing acceptance |
| Class 1 IVD medical devices do not require manufacturer evidence | At any time, the TGA may request documents to demonstrate a class 1 IVD complies with the requirements. |
What sponsors must do to transition to new evidence
The transition from one form of manufacturer evidence to another may result in the sponsor needing to notify the TGA of additional changes or apply to the TGA for regulatory approvals.
For example, the manufacturer’s new certification may cover a different scope of activities or kinds of device or limit the intended purpose of the IVD covered by the certificate. The new certification could also be associated with a change that triggers automatic conditions of inclusion that require certain changes to be notified to the TGA.
The following sections outline the possible actions sponsors must consider when transitioning IVD ARTG entries to new manufacturer evidence. Examples are included below each section.
- Updating manufacturer evidence
- Lapsing or expiring manufacturer evidence
- Change in classification due to change in intended purpose
- Change to ARTG entry information
- Non-compliance with the Essential Principles
- Change in device safety and performance
- Change to intended purpose of a device without change in classification
- Supplying a new device under a current ARTG entry
Note
At all times, an IVD medical device sponsor must:
- maintain ARTG entries that cover the IVD medical devices they supply
- maintain manufacturer evidence that supports their ARTG entries.
The scope of any new manufacturer evidence must therefore cover the IVD medical devices included under the relevant ARTG entry.
Updating manufacturer evidence: section 1
| Updating manufacturer evidence | Sponsor action |
|---|---|
Update the manufacturer evidence associated with your ARTG entries to your new certification.
| You can update your manufacturer evidence on file with the TGA, to your new certification, using one of the following pathways:
For further information see: Manufacturer evidence for medical devices. |
Case study 1: Updating manufacturer evidence
BlueDx is the sponsor of two COVID-19 IVD devices under one ARTG entry. The ARTG entry is currently supported by ISO 13485 certification. The sponsor wishes to update to new MDSAP certification. The MDSAP certificate covers the same two devices covered by the ISO 13485 certificate.
BlueDx will need to submit a variation to manufacturer evidence.
Additional examples and scenarios:
Example 1: Multiple manufacturer evidence certificates for one ARTG entry
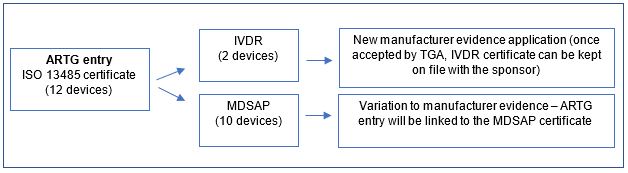
A sponsor has an existing ISO 13485 certificate that covers all 12 devices listed under a single ARTG entry. The sponsor is transitioning to new MDSAP certification (covers 10 devices) and new IVDR certification (covers 2 devices), where neither certificate fully covers the range of existing devices.
The sponsor submits a variation to manufacturer evidence application for the MDSAP certificate (to replace the existing ISO 13485 certificate), as it covers most devices under the ARTG entry. This will link the ARTG entry to the MDSAP certificate.
The sponsor also submits a new manufacturer evidence application for the IVDR certificate to support the inclusion of the remaining 2 devices.
Once the TGA accepts this certificate as new manufacturer evidence, the sponsor can keep the IVDR certificate on file as evidence of conformity assessment.
Example 1 flow chart
{kind=link}
A flow chart showing the certification process starting with 'ARTG entry ISO 13485 certificate (12 devices)' on the left.
This branches into two paths: The upper path shows 'IVDR (2 devices)' leading to 'New manufacturer evidence application (once accepted by TGA, IVDR certificate can be kept on file with the sponsor)'.
The lower path shows 'MDSAP (10 devices)' leading to 'Variation to manufacturer evidence - ARTG entry will be linked to the MDSAP certificate'. Each box is connected by arrows indicating the flow direction."
Example 2: DCR to update only some of your ARTG entries associated with the same evidence
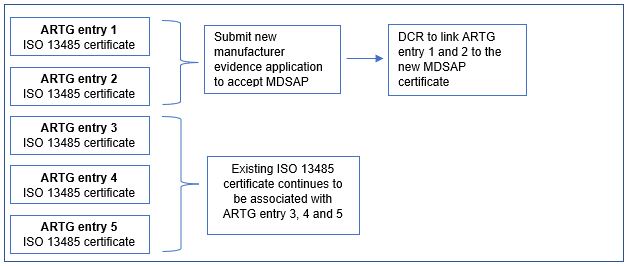
A sponsor has 5 ARTG entries associated with an ISO 13485 certificate all linked to the same manufacturer evidence identifier. Two of the 5 ARTG entries are covered by a new MDSAP certificate and can be updated.
A new manufacturer evidence application for the MDSAP certificate can be submitted, followed by a DCR to link these 2 ARTG entries from the ISO 13485 certificate to the new MDSAP certificate. The 3 remaining ARTG entries will continue to be linked to the ISO 13485 certificate and will need to be updated once the ISO 13485 certificate expires.
Example 2 flow chart
{kind=link}
A flowchart diagram showing the ARTG entry and ISO certification process. On the left side are five ARTG entries (1 through 5), each with an associated ISO 13485 certificate.
These entries connect to two central processes: The top process shows 'Submit new manufacturer evidence application to accept MDSAP' which leads to 'DCR to link ARTG entry 1 and 2 to the new MDSAP certificate'.
Below this, there's a note stating 'Existing ISO 13485 certificate continues to be associated with ARTG entry 3, 4 and 5'.
All elements are connected by simple lines indicating the flow of the process.
Example 3 Changing the manufacturer evidence for a class 4 IVD
New applications and existing ARTG entries for class 4 IVDs can be supported by the following manufacturer evidence and associated design examination certification:
- TGA conformity assessment certificate
- Certification issued under EU IVDD
- Certification issued under EU IVDR
- MDSAP supported by Health Canada medical device license
- US FDA PMA
- Premarket approval by Japan PMDA
- Approval from HSA Singapore
A sponsor of a class 4 IVD ARTG entry currently supported by TGA conformity assessment and design examination, would like to transition all devices to certification issued under EU IVDR. The sponsor can submit a variation to manufacturer evidence to update the evidence and must notify the TGA that they will not be seeking recertification of the TGA conformity assessment certificate.
Lapsing or expiring manufacturer evidence: section 2
| Change | Sponsor action |
|---|---|
Lapsing refers to any loss or gap in certification (i.e., a period where the sponsor no longer holds valid manufacturer evidence to support their ARTG entry); or if there is a loss in certification for some of the devices covered under an existing ARTG entry. At all times, a sponsor must have available sufficient information to substantiate conformity assessment procedures have been applied to the kind of medical device in the ARTG. | IVD medical devices included in the ARTG that were manufactured under valid conformity assessment evidence can be supplied after the evidence has expired. Sponsors should notify the TGA within 60 days of becoming aware of the lapsing, revocation, suspension or cancellation of conformity assessment evidence (manufacturer evidence) using the notification form for Lapses in Medical Device Conformity Assessment Certification. The TGA may review the information provided to determine whether the entries can remain included in the ARTG. Any decision made is case-by case. There are criminal and civil penalty sanctions if a sponsor fails to notify the TGA within 60 days of becoming aware that manufacturer evidence (other than a TGA conformity assessment certificate) has been restricted, suspended, revoked or is no longer in effect. Further information can be found in TGA conformity assessment guidance. |
Case study 2: Loss or gap in certification for an ARTG entry
AllDx is the sponsor of a class 3 IVD ARTG entry supported by ISO 13485 certification and the manufacturer has applied for IVDR certification. The notified body has not issued the new IVDR certificate before the ISO 13485 certificate expires. This means there will be a gap in valid certification to support the device supplied under the ARTG entry.
AllDx will need to submit a notification form for lapses in medical device conformity assessment certification, within 60 days. The TGA will then advise AllDx on the appropriate next steps.
Additional examples and scenarios:
Example 4: No loss in certification for an ARTG entry
If there is no loss or gap in valid certification, and no change in the scope of the devices covered (that is, the new certification overlaps with or comes into effect immediately after the old certification expires) you do not need to notify the TGA via a notification form for lapses in medical device conformity assessment certification.
Instead, you may wish to update your ARTG entry by completing a variation to manufacturer evidence application, provided the intended purpose and use of the device is the same, and there are no changes to the device safety or performance.
For example, a sponsor is transitioning from ISO 13485 to MDSAP certification to support their ARTG entry. The new MDSAP certificate is issued before the ISO 13485 certificate expires and the scope covers all the same devices.
The sponsor does not need to advise the TGA that the ISO 13485 certificate is expiring as the ARTG entry will be covered by the MDSAP certification. The sponsor may instead wish to update the entry with the new certification via a variation to manufacturer evidence application.
Example 5: Loss in certification for a device
A reduction in the scope of new certification that will be used to support an existing ARTG entry may require a notification form for Lapses in medical device conformity assessment certification to be submitted to the TGA if some of the devices are no longer covered by valid QMS conformity assessment certification (manufacturer evidence).
For example, a sponsor is transitioning from ISO 13485 to certification under EU IVDR to support the 10 devices under their existing ARTG entry. The certification under EU IVDR comes into effect immediately following the expiry of the ISO 13485 certificate.
The scope of the EU IVDR certification now only covers 6 of the devices currently supplied under the ARTG entry. This means there is a gap in certification for the 4 devices not covered by the scope of the certificate issued under EU IVDR. A notification form for lapses in medical device conformity assessment is required to be submitted and the TGA will advise on the appropriate action.
Change in classification due to change in intended purpose: section 3
| Change | Sponsor action |
|---|---|
| Potential changes to the scope of indications or intended purpose which could change the classification for the IVD medical device under the Australian regulatory framework. | You will need to lodge a new application for inclusion in the ARTG for your affected IVD medical device (unless you have an existing alternative ARTG entry). The type of manufacturer evidence used to support a new application for inclusion and the intended purpose of the device, will determine if an IVD application is selected for mandatory audit. Regardless of the type of certification some applications may be selected for non-mandatory audit at the TGA’s discretion. |
Example 6: Change in IVD class from class 2 to class 3 IVD companion diagnostic
In the process of transitioning from ISO 13485 to certification issued under EU IVDR, a manufacturer determines one of their existing immunohistochemistry IVDs will now be intended to be used as a companion diagnostic.
The EU notified body issues a certificate covering the device for this purpose. As the device is currently included in the ARTG as a class 2 IVD, the sponsor will need to submit a new IVD application for a class 3 IVD companion diagnostic (before the end of the companion diagnostic transition period) to continue supplying the device.
As the device is supported by certification issued under EU IVDR, it will not be selected for mandatory audit but may be selected for non-mandatory audit. Further actions to notify users and update labelling for old stock may also be needed.
Change to ARTG entry information: section 4
| Change | Sponsor action |
|---|---|
Changes to any of the following:
Note 1: GMDN code refers to collective terms (class 1-3 IVDs) and preferred terms (class 4 IVDs). Change in GMDN code cannot change the ‘kind of device’, otherwise a new IVD application is required. Note 2: If the manufacturer’s legal entity changes, or products are transferred to a new manufacturer, a new application for ARTG inclusion is required. | You can update your ARTG entries by submitting a Device Change Request (DCR) application for your affected ARTG entries. The same process applies regardless of the type of manufacturer evidence used to support the entry, except TGA conformity assessment (see below). If the same change is being applied across multiple ARTG entries, you can update up to 10 ARTG entries in one DCR application. An additional DCR application would be needed if more than 10 ARTG entries required the same change.
TGA Conformity AssessmentIf an ARTG entry is supported by a TGA conformity assessment certificate and there are substantial changes to the design of the device that impact the device quality, safety and performance, an application to change the TGA conformity assessment certificate will be required. Further information on Device Change Request and variation applications can be found in Varying entries in the ARTG. |
Case study 3: Changing the intended purpose of an ARTG entry
BestDx is the sponsor of a Class 3 IVD ARTG entry that covers both self-tests and point of care tests supported by ISO 13485 certification.
The sponsor has transitioned to IVDR certification that covers point of care tests only. The sponsor has no other certification or evidence to support the supply of self-tests under the ARTG entry.
BestDx will need to submit a Device Change Request to update the ARTG entry to reflect that only point of care tests are approved.
Additional examples and scenarios
Example 7: Changing GMDN code
A sponsor identifies a more preferred GMDN code for their existing ARTG entry that does not change the kind of device (a change in ‘kind of device’ would require a new ARTG inclusion application). A Device Change Request can be submitted to change the GMDN code stated in their ARTG entry.
Example 8: Changing the information included in the ARTG for a class 4 IVD supported by EU IVDR versus TGA conformity assessment
Regardless of the type of certification, a sponsor must notify the TGA of any changes to the class 4 IVD device resulting in a change to the information included in the ARTG (for example, a change in the device that subsequently results in a change to the ARTG intended purpose). If the ARTG entry is supported by:
- Certification issued under EU IVDR: Only changes to the device or manufacturer that will result in a change to the information displayed in the ARTG entry must be notified to the TGA via a Device Change Request.
- if an additional condition of inclusion has been imposed on an ARTG entry, the sponsor must comply with this condition and provide relevant information to the TGA for assessment
- sponsors must keep all relevant records related to device changes and make them available to the TGA upon request.
- TGA Conformity Assessment: A change to TGA conformity assessment certificate application is required for substantial changes to the design of the device that impact the device quality, safety and performance.
Non-compliance with the Essential Principles: section 5
| Change | Sponsor action |
|---|---|
The transition to certification under EU IVDR or other manufacturer evidence may require sponsors to request consent to import, supply or export IVD medical devices in their control which do not meet an Essential Principle for a limited period. Examples may include (but are not limited to):
| If you determine that your IVD medical devices, which are manufactured under a valid manufacturer evidence certificate, no longer comply with the Essential Principles for safety and performance, an application for consent must be submitted before importing, exporting, or supplying the devices. A risk mitigation plan outlining how the non-compliance will be addressed and notified to the end user is required. To complete a consent application, the sponsor needs to complete the online application form available on the TGA Business Services (TBS) portal. Further information on submitting a consent application and associated fees is available in TGA guidance. Where consent to supply is granted for non-compliance with the Essential Principles, ongoing regulatory responsibilities of the sponsor remain including, but not limited to, undertaking recall actions, and the reporting of adverse events. The TGA conducts post-market reviews to ensure that devices in the ARTG continue to meet legislative obligations. There are criminal and civil penalty sanctions if a sponsor imports, exports, or supplies non-compliant IVD medical devices without consent. Note: Some fee concessions apply for applications for consent made solely in relation to non-compliance with Essential Principle 13 due to transitioning certification from the EU IVDD or ISO 13485 to EU IVDR or MDSAP. |
Example 9: Consent to Supply for use of a sample type no longer supported by certification
A sponsor is transitioning from ISO 13485 certification to MDSAP certification, to support inclusion of their Sexually Transmitted Infection (STI) test in the ARTG. The MDSAP certificate has been issued with a reduced scope that no longer covers use of the test with urine samples and is now valid for use with swab only.
If the sponsor wishes to continue supplying the old stock manufactured while the ISO 13485 certification was valid (while also supplying the new stock manufactured under the new MDSAP certification) consent to supply will be required if the manufacturer does not hold the clinical and analytical evidence to support use of the ISO 13485 stock with both urine and swab samples; and the IFU has not been updated to reflect the change. The device is non-compliant with Essential Principle 13, 14 and 15.
Change in device safety and performance: section 6
| Change | Sponsor action |
|---|---|
A change in safety and performance of a device that has been supplied to market.
| Device safety and performance issues that arise through the normal use of your devices which have been supplied to the market must be notified to the TGA. The Recalls Section in the TGA will discuss with you, the appropriate action. These actions may include recall or non-recall actions. Recall actions are typically undertaken in accordance with the Uniform Recall Procedure for Therapeutic Goods (URPTG) and occur regardless of the type of manufacturer evidence supporting the devices under an ARTG entry. See Manage a recall for further information on recall and non-recall actions. |
Change to intended purpose of a device without change in classification: section 7
| Change | Sponsor action |
|---|---|
Change to the intended purpose of a device under a current ARTG entry. For example, to broaden or reduce the indications for use; intended user or target patient population. If the device classification and ARTG entry details are unchanged, you only need to notify the TGA in limited cases. | Certification under EU IVDRFor EU IVDR certification, you do not need to notify the TGA about changes to the intended purpose that do not change the classification or details in the ARTG entry if that entry is supported by certification under EU IVDR. TGA conformity assessmentFor TGA conformity assessment, if an ARTG entry is supported by a TGA conformity assessment certificate and there are substantial changes to the design of the device that impact the device quality, safety and performance, an application to change the TGA conformity assessment certificate will be required. Other certification (e.g., EU IVDD, MDSAP or ISO 13485)For other certification (e.g., EU IVDD, MDSAP or ISO 13485), for certain devices (see Appendix A) with non-TGA and non-EU IVDR certification, automatic conditions of inclusion require changes to the intended purpose of a device to be notified to the TGA via a Device Change Request (DCR). Additional ActionsRegardless of the type of manufacturer evidence used to support the ARTG entry, a change to the intended purpose of a device (e.g., reduced indications for use) may require recall action to notify the market and end users of the change. If there will be a delay in updating the labelling and instructions for use to reflect the change, you may need to submit an application for Consent to supply medical devices that are non-compliant with the Essential Principles. |
Example 10: Changes to the intended purpose of a device
A sponsor is updating the intended purpose of a point of care device currently included in the ARTG to add a new self-test for a class 3 IVD device. If the ARTG entry is supported by:
Certification issued under EU IVDR:
- a Device Change Request is only required if the information in the ARTG certificate needs updating (for example, changing the ARTG intended purpose)
- sponsors must keep all relevant records related to changes to the intended purpose of a device and make them available to the TGA upon request
- if an additional condition of inclusion has been imposed on an ARTG entry, the sponsor must comply with this condition and provide relevant information to the TGA for assessment.
For other certification such as certification issued under EU IVDD, MDSAP or ISO 13485 certification:
- a variation is needed to add a new device of the same kind and change the intended purpose as notification to the TGA is required under automatic conditions of inclusion (see Appendix A)
- data specific to self-test may be reviewed at this time.
Supplying a new device under a current ARTG entry: section 8
| Change | Sponsor action |
|---|---|
Adding a new device of the same kind under a current ARTG entry. Change in product name or intended purpose, due to addition of a new device of the same kind under a current ARTG entry requires notification in limited cases. | Class 4 IVDs and companion diagnostics:
EU IVDR certification:
Other certification (e.g., EU IVDD, MDSAP or ISO 13485)
Further information on variation applications can be found in Varying entries in the ARTG. |
Case study 4: Changing the intended purpose of an ARTG entry
BlueDx is the sponsor of an ARTG entry of a class 3 nucleic acid test to detect Chlamydia. The ARTG entry is supported by certification issued under EU IVDR with no additional conditions of inclusion imposed.
The sponsor would like to supply another Chlamydia antibody test that is the same kind of device and is covered by the scope of the devices listed under the certificate issued under EU IVDR.
BlueDx does not need to notify the TGA or submit a variation application to supply the additional device under the ARTG entry.
Example 11: MDSAP or ISO 13485 certification and adding a new device of the same kind (in Appendix A)
A notification to the TGA (variation application) is required to supply an additional device of the same kind for certain devices (see Appendix A), if the ARTG entry is supported by other certification such as MDSAP or ISO 13485 certification.
For example, a sponsor has an existing ARTG entry for a class 3 IVD nucleic acid test to detect Chlamydia, supported by MDSAP or ISO 13485 certification.
To supply an additional Chlamydia antibody test (with different product name) that is considered the same kind of device under the ARTG entry, a notification to the TGA is required as part of the condition of inclusion.
Example 12: Adding a new class 3 IVD device of the same kind (not in Appendix A)
If an ARTG entry is supported by certification under EU IVDD, MDSAP or ISO 13485 certification, an IVD variation is not required to supply the class 3 IVD device of the same kind, if the additional device:
- is not specified in Appendix A, and
- supported by acceptable evidence of product review (for example, Health Canada Class III medical device licence or US FDA PMA).
Examples of class 3 IVDs (not specified in Appendix A) that would not require an IVD Variation application to supply under an existing ARTG entry (if supported by certification issued under EU IVDD, Health Canada Class III medical device licence or US FDA PMA as evidence of product review) include tests for (but not limited to):
- Trisomy 21
- Phenylketonuria
- Human Leucocyte Antigen
- Prostate Specific Antigen.
Example 13: When a sponsor with certification issued under EU IVDR may need to notify the supply of an additional device
If an ARTG entry is supported by certification issued under EU IVDR, notification to the TGA is not usually required to supply an additional device of the same kind, unless additional conditions of inclusion have been imposed or ARTG details need to be changed.
For example, a sponsor submits an IVD Application for inclusion for a point of care test that is supported by certification issued under EU IVDR. At the time of approval, an additional condition of inclusion is imposed in the ARTG entry requiring the sponsor to notify the TGA if a self-test (which is the same kind of device) is to be supplied under this ARTG entry. The sponsor must submit an IVD variation application if they intend to supply a self-test under the entry.
Example 14: Adding a new class 4 immunohematology reagent (IHR) under an existing ARTG entry (as the same kind of device)
A sponsor would like to supply two new class 4 IHR devices under an existing ARTG entry. As class 4 IHR devices do not have a unique product identifier requirement, the additional devices of the kind, will need notification to the TGA as it changes the details included in the ARTG, a Device Change Request would be required to update the ARTG certificate.
Note: All other class 4 IVDs (except IHR) would require a new class 4 application for inclusion to be submitted to the TGA.
Appendix A: IVDs with conditions of inclusion to notify the TGA
Note
On 1 July 2024, amendments to Regulation 5.12 of the Therapeutic Goods (Medical Devices) Regulations 2002 came into effect.
This changed the types of IVD medical device ARTG entries for which the sponsor must notify the TGA about changes to the intended purpose or about the supply of new devices of the same kind.This Appendix provides guidance relevant to ARTG entries approved before 1 July 2024.
We are preparing new guidance that reflects the 1 July 2024 amendments, and this will be published soon. We thank you for your patience while we prepare the updated guidance.
If you have any question regarding the obligations of sponsors under Regulation 5.12, email ivds@tga.gov.au and we will respond as quickly as possible.
Some IVDs have automatic conditions of inclusion that require the sponsor to notify the TGA about changes to the intended purpose or about new devices of the same kind to be supplied under that ARTG entry.
This does not apply to IVDs supported by certification issued under EU IVDR or TGA conformity assessment certification. This applies to a limited subset of IVDs (see Table 2) with non-TGA conformity assessment and non-EU IVDR certification.
Note
Regulation 5.12 is an automatic condition of inclusion, which specifies that the supply of a new device of the kind under an existing ARTG entry, that results in change of product name or change to the intended purpose requiring notification to the TGA.This condition of inclusion only applies to devices specified under Regulation 5.3(1)(j). See Table 2.
This does not apply for devices supported by certification issued under EU IVDR or TGA conformity assessment certificate.
| Regulation 5.3(1)(j) | IVDs with conditions of inclusion to notify the TGA | Example |
|---|---|---|
| (i) | Quality Control material for a class 4 IVD | Controls for ABO blood typing |
| (ii and iii) | Self-tests and point of care tests | COVID-19 Rapid Antigen tests |
| (iv) | Class 3 IVDs for sexually transmitted infections | Chlamydia and Gonorrhoea tests |
| (v) | IVDs for managing or monitoring the treatment of infections diagnosed using a class 4 IVD medical device | Viral load tests for Human Immunodeficiency Virus and Hepatitis C Virus |
| (vi and vii) | IVDs intended to be supplied for use in Pharmaceutical Benefits Scheme or use in a national screening program | Faecal Occult Blood Screening Program tests |
| (viii) | A device where the Secretary is not satisfied whether appropriate evidence of conformity assessment is held to demonstrate evidence of product review | Class 3 IVDs (e.g., prenatal screening and CD cell cancer biomarker IVDs) supported by ISO 13485 certification without additional evidence of product review (e.g., Health Canada Class III licence) |
| (viii)(a) | Class 4 IVDs | ABO blood typing tests |
| (x) | IVD companion diagnostics | IVDs that provide information essential for the safe and effective use of a corresponding medicine or biological |
Even when these conditions of inclusion do not apply, if the supply of an additional device of the same kind or a change to the intended purpose of an existing device results in changes to the information in the ARTG, a DCR is needed to update the ARTG certificate.
There may also be other instances where additional conditions are imposed on an ARTG entry requiring a sponsor to notify of any additional devices to be supplied.
Note
The ARTG certificate only displays additional devices that are notified to the TGA in a variation application.
Additional case studies and scenarios
We have provided additional case studies and scenarios about manufacturer evidence for sponsors transitioning to new conformity assessment certification.
Many sponsors and manufacturers of In Vitro Diagnostic (IVD) medical devices will be seeking to transition to new manufacturer evidence because of either:
- The new European Union (EU) IVD Regulation 2017/746 (EU IVDR) that replaces EU IVD Directive 98/79/EC (IVDD).
- The TGA phase out of acceptance of many ISO 13485 certificates as manufacturer evidence1, unless supported by manufacturer’s EU Declaration of Conformity (DoC) made under the IVDD prior to 26 May 2022.
This guidance is to be read in conjunction with Transition to new manufacturer evidence for IVD medical devices.
Case Studies
Case study: Michael
Michael is an applicant who intends to make an application for inclusion for a Class 3 IVD. He is using an active ISO 13485 certificate issued by a Notified Body designated under the EU IVDD as supporting conformity assessment evidence.
Michael will need to provide an alternative certification (see Therapeutic Goods (Medical Devices—Information that Must Accompany Application for Inclusion) Determination 2018) acceptable for Class 3 IVD or provide EU DoC issued by the manufacturer under EU IVDD before 26 May 2022 to continue with the ARTG inclusion application.
TGA no longer accepts ISO 13485 certificates issued by a Notified Body designated under the EU IVDD as conformity assessment evidence for new ARTG inclusion applications, unless supported by EU DoC.
Case study: Craig
Craig is an applicant who intends to make an application for inclusion for a Class 3 IVD. He is using an active ISO 13485 certificate issued by an accreditation body that is a signatory to the IAF MLA as supporting conformity assessment evidence.
Craig will need to provide an alternative certification (see Therapeutic Goods (Medical Devices—Information that Must Accompany Application for Inclusion) Determination 2018) acceptable for Class 3 IVD or provide EU declaration of conformity issued by the manufacturer under EU IVDD before 26 May 2022 to continue with the ARTG inclusion application.
TGA no longer accepts ISO 13485 certificates issued by a signatory to the IAF MLA as conformity assessment evidence for new ARTG inclusion applications, unless supported by EU DoC.
Case study: Amanda
Amanda has an ongoing ARTG inclusion application submitted before 26 May 2023 and supported by an ISO 13485 certificate that expired during the application process.
Amanda is required to submit an updated ISO 13485 certificate or an alternative form of evidence (see Therapeutic Goods (Medical Devices—Information that Must Accompany Application for Inclusion) Determination 2018) before a decision can be made on the application.
TGA can progress the assessment of the application, but it will not be able to make a decision without valid conformity assessment evidence at the time of decision.
Case study: Robert
Robert is a sponsor who has an ARTG entry for a Class 3 IVD supported by a valid ISO 13485 certificate and the details have been included in the TGA records.
No action is required at this point as the sponsor can continue to supply devices already included in the ARTG, till supported by valid ISO 13485 certificate.
Robert is encouraged to transition to an alternative form of evidence, noting the policy intent to eventually phase out the acceptance of ISO 13485 certificates for existing ARTG entries.
Case study: Paul
Paul is a sponsor who has an ARTG entry for a Class 2 IVD supported by a valid ISO 13485 certificate but has not updated these details in the TGA records. The TGA records only has details of an expired ISO 13485 certificate.
Paul can continue to supply devices already included in the ARTG and is recommended to submit a Manufacturer evidence variation to update the TGA records.
Paul is encouraged to transition to alternative form of evidence before the ISO certificate expires, noting the policy intent to eventually phase out the acceptance of ISO 13485 certificates for existing ARTG entries.
Case study: Nick
Nick is a sponsor who has an ARTG entry for a Class 3 IVD which is supported by an expired ISO 13485 certificate. The manufacturer does not have a valid ISO 13485 certificate or any other form of acceptable certification. The TGA records only has details of the expired certificate.
Nick can continue to supply products that were manufactured when the ISO 13485 certificate was valid, but will need to obtain a new form of valid acceptable evidence (see Therapeutic Goods (Overseas Regulators) Determination 2018) from the manufacturer to continue to supply in Australia any products manufactured after the certification expired.
Nick must notify the TGA within 60 days of becoming aware that the ISO 13485 certificate expired using the notification form for lapses in medical device conformity assessment certification.
Case study: Jenny
Jenny is a sponsor who has an ARTG entry for a Class 4 IVD that is transitioning from EU IVDD to EU IVDR certification. During this process, the IVDD Design Certificate supporting their Class 4 IVD device has expired.
For a Class 4 IVD, the Therapeutic Goods (Medical Devices—Information that Must Accompany Application for Inclusion) Determination 2018 states the conformity assessment evidence related to manufacturers Quality Management System along with Design examination (i.e. product assessment evidence) is required.
Jenny can continue to supply products that were manufactured when the IVDD certificate was valid, but will need to obtain a new form of valid acceptable evidence (see Therapeutic Goods (Overseas Regulators) Determination 2018) from the manufacturer to continue to supply in Australia any products manufactured after the certification expired.
Jenny must notify the TGA within 60 days of becoming aware that the certificate expired using the notification form for lapses in medical device conformity assessment certification.
Scenarios for IVD ARTG entries and applications
1.What if the scope of the new EU IVDR certificate has changed and the change no longer covers some of the devices that are supplied under the ARTG entries?
The sponsor can continue to supply all products that were manufactured under the previous valid certification, but will need to obtain a new form of valid acceptable evidence (see Therapeutic Goods (Overseas Regulators) Determination 2018) to continue to supply in Australia the products that are not covered under the new IVDR certification.
They must notify the TGA within 60 days of becoming aware that the certificate lapsed/expired using the notification form for lapses in medical device conformity assessment certification.
If the sponsor is unable to obtain alternative certification and if the scope of the manufacturer evidence certificate no longer supports all the devices included in the ARTG entries, they must submit a request to the TGA to cancel the relevant ARTG entries not covered by the conformity assessment evidence.
The sponsor is encouraged to contact TGA at devices@tga.gov.au if they wish to discuss any impact on supply disruptions.
2. I have submitted an inclusion application with ISO 13485 certificate issued before 26 May 2023 as manufacturer evidence. The ISO certificate expires in two months from the application date. Can I submit a renewed certificate later in the application process?
Yes, the sponsor will need to provide the TGA either with an updated ISO 13485 certificate or a new form of acceptable conformity assessment certification for the application to be progressed. The TGA will not be able to progress an application without a valid manufacturer evidence, at the time of decision.
3. I wish to apply for inclusion of Class 2 IVDs in Australia. The manufacturer is transitioning to EU IVDR certification. However, these devices are lower class (i.e., Class A) under EU IVDR regulations. What is the evidence required for the purposes of TGA application?
Class A devices in Europe are self-certified. As the devices are classified as Class 2 IVD in Australia, you will be required to provide an acceptable form of certification for Class 2 IVDs. Refer to Therapeutic Goods (Medical Devices—Information that Must Accompany Application for Inclusion) Determination 2018 for acceptable conformity assessment evidence.
4. Can I submit an ARTG inclusion application with Class 2 IVD supported by ISO 13485 certificate issued by either an accreditation body that is a signatory to the IAF MLA or a Notified Body designated under the EU IVDD?
Yes, a class 2 IVD inclusion application can be submitted supported by ISO 13485 certificate issued by either an accreditation body that is a signatory to the IAF MLA or a Notified Body designated under the EU IVDD as conformity assessment evidence, if it is associated with the manufacturer’s European Declaration of Conformity made under the EU IVDD before 26 May 2022. This is acceptable until 26 May 2027 for Class 2 IVDs.
Footnotes
- The TGA continues to recognise ISO 13485 as an acceptable standard. However, certificates issued by either of the following bodies, are no longer accepted as manufacturer evidence for new IVD inclusion applications unless accompanied by manufacturer’s EU DoC made prior to 26 May 2022.
- A body accredited by a signatory to the Multilateral Recognition Arrangement of the International Accreditation Forum (IAF MLA), or
- A Notified Body designated under the EU IVDD.
Page history
Updates to the guidance:
- Change in line with further extension of EU IVDR timelines
- Changes to Regulation 5.3 of Therapeutic Goods (Medical Device) Regulations 2002.
Title changed from 'Transition to new manufacturer evidence for IVD medical devices' to 'Transitioning to new manufacturer evidence for in-vitro diagnostic medical devices (IVDs)' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Publication of 'additional case studies and scenarios'.
Original publication.
Updates to the guidance:
- Change in line with further extension of EU IVDR timelines
- Changes to Regulation 5.3 of Therapeutic Goods (Medical Device) Regulations 2002.
Title changed from 'Transition to new manufacturer evidence for IVD medical devices' to 'Transitioning to new manufacturer evidence for in-vitro diagnostic medical devices (IVDs)' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Publication of 'additional case studies and scenarios'.
Original publication.