Understanding rules for manufacturing biologicals
Guidance on TGO 109: Standards for Biologicals - General and Specific Requirements.
Purpose
This guidance is for manufacturers and sponsors of biologicals. It specifies the general manufacturing and quality requirements for therapeutic goods comprising, derived from or containing human cells or tissues (HCT) set out in the Therapeutic Goods (Standards for Biologicals-General and Specific Requirements) (TGO 109) Order 2021 and provides an interpretation of how the various requirements can be met
Generally, Therapeutic Goods Orders (TGOs) are standards that determine the consistency of product quality, including label quality. As an HCT product provider, you must comply with requirements that contribute to the quality and safety of HCT products, and that mitigate infectious disease risks.
TGOs are subject to automatic repeal 10 years after their registration (sunsetting). TGO 109 replaced the following TGOs:
- Therapeutic Goods Order No. 83 - Standards for human musculoskeletal tissue (TGO 83)
- Therapeutic Goods Order No. 84 - Standards for human cardiovascular tissue (TGO 84)
- Therapeutic Goods Order No. 85 - Standards for human ocular tissue (TGO 85)
- Therapeutic Goods Order No. 86 - Standards for human skin (TGO 86)
TGO 109 also includes some aspects of this TGO:
TGO 88 does not sunset until 1 October 2023 but was also updated given the crossover with the product-specific TGOs 83-86.
This information is provided for guidance only and has been developed based on current knowledge of the subject matter.
It should not be relied on to address every aspect of the relevant legislation. You should seek your own independent legal advice to ensure that all legislative requirements are met. For clarification of a particular requirement, contact TGA’s Biological Science Section (BSS).
Read this guidance in conjunction with TGO 109.
Legislation
About TGO 109
The Therapeutic Goods (Standards for Biologicals-General and Specific Requirements) (TGO 109) Order 2021 specifies minimum manufacturing and quality requirements including:
- general standards for all biologicals (Part 2) specific standards for human tissues which have only been subjected to minimal manipulation:
- musculoskeletal products (Part 3)
- cardiovascular products (Part 4)
- ocular products (Part 5) skin products (Part 6)
- amniotic products (Part 7).
TGO 109 applies to:
- all biologicals (encompassing HCT materials, starting materials, intermediates and finished therapeutic goods) that come within the operation of Part 3-2A of the Therapeutic Goods Act 1989 (the Act).
TGO 109 does not apply to:
- biologicals that are not intended for therapeutic use, that is, blood or tissue samples for infectious disease or bioburden testing
- exempt autologous HCT products biologicals mentioned in item 13 of Schedule 5A to the Therapeutic Goods Regulations 1990 (the Regulations), subject to compliance with the condition specified in that item.
If you are unsure whether TGO 109 applies to a specific biological, please contact TGA’s Biological Science Section (BSS).
Review of TGO 109
TGO 109 will be reviewed regularly as changes in legislation, emerging technology, and best practice occur. Sponsors and manufacturers are encouraged to discuss with TGA any proposed changes in practice or evolving technologies that may affect, or be affected by, the requirements of TGO 109.
Guidance on the requirements of TGO 109
Allogeneic use in relation to an HCT product means administration to, or application in the treatment of, a person other than the person from whom the HCT materials used in the manufacture of the product were collected.
For more information on Minimal manipulation, see:
These specifications were in part guided by requirements in the Council of Europe’s 4th Edition of the Guide to the quality and safety of tissues and cells for human application (2019).
Part 2 - Standard for all biologicals
Section 8 - What this Part is about
8 What this Part is about
This Part specifies general requirements that must be met in the manufacture of all biologicals, including requirements relating to the HCT materials used in the manufacture of those biologicals.
Unless otherwise specified, the requirements in this Part apply to all biologicals, including a biological that is subject to a specific standard in Parts 3 to 7 of this instrument.
This includes HCT collected and/or in various stages of processing for manufacture of the biological.
Section 9 – Diseases and conditions that may compromise biologicals
9 Diseases and conditions that may compromise biologicals
- A biological must not be manufactured using HCT materials collected from a donor who is known to have a disease or condition that may compromise the quality, safety or efficacy of the biological, unless:
- criteria for donor selection and periods of donor ineligibility, based on validated data or documented evidence from relevant scientific literature, are applied that support and justify the quality, safety and efficacy of the biological, and the application of the criteria is documented; or
- in any case where it is not possible to comply with paragraph (a)—a registered medical practitioner acting for or on behalf of the manufacturer or the sponsor has agreed to, and documented the rationale for, the use of the HCT materials in the manufacture of the biological.
Some diseases, pathological conditions and infectious diseases, might affect the quality, safety or efficacy of the biological. Donor age, medical treatment prior to collection may or infectious diseases mentioned in TGO 108 may adversely affect the quality of the HCT material or the biological.
The criteria for donor selection depend on the type and the intended use of the biological and could include factors such as:
- tissues exposed to irradiation prior to collection
- diseases that affect joint integrity in musculoskeletal donors
- immunodeficiency disorders or the use of immunosuppressive agents in patients with a consequent higher risk of asymptomatic opportunistic infection
- reactivation of past infection
- situations that could impair the ability of testing to detect infection, for example, hypogammaglobulinaemia, HCV antibody testing.
Sponsors must specify additional criteria that may be applicable and this should be based on clinical justification. Ensure that relevant deferral criteria are communicated to organisations conducting the collection process, to ensure donors are screened for these criteria before acceptance of the starting material.
Consult relevant tissue-specific chapters of the Council of Europe’s 4th Edition of the Guide to the quality and safety of tissues and cells for human application (2019). to establish an initial list of conditions that should result in deferral or rejection of donors of specific tissue types. These lists are not exhaustive, and the manufacturer should conduct their own risk assessment to develop and maintain donor selection criteria based on their HCT product and manufacturing process.
Manufacturers must consider what testing is required to ensure the safe manufacture and use of the HCT product. Justification for the selection and evaluation of disease and conditions should be included in submitted dossier.
A registered medical practitioner may accept donors with underlying disease or conditions if they have performed an adequate risk assessment/have an adequate rationale considering the intended use of the HCT product and the intended recipient. Provide the justification/risk assessment in the relevant section of the dossier.
Section 10 - Critical materials
10 Critical materials
- The critical materials used in the manufacture of a biological must:
- not be contaminated with, or be likely to introduce, microorganisms or other infectious disease agents; and
- not adversely affect the quality, safety or efficacy of the biological; and
- where the critical materials are solutions that come into contact with HCT materials or biologicals (other than critical materials mentioned in paragraph (d))—be manufactured under an approved quality management system and either:
- be supplied as a sterile solution; or
- satisfy sterility requirements specified in an applicable standard; and
- where the critical materials are non-sterile antimicrobial agents used in a bioburden reduction process for HCT materials—be passed through a 0.22µm filter prior to use in the bioburden reduction process; and
- where the critical materials are of human or animal origin (other than HCT materials)—meet the requirements set out in:
- the TGA Approach to TSE; and
- the Guidance on Virus Validation Studies.
Critical materials not included in the Register
- The following information in relation to critical materials that are not included in the Register must be available and documented:
- screening tests performed and quality control specifications, including criteria and limits for tests; and
- storage conditions.
Critical materials include all supplies, reagents, primary containers and collection kits that come into direct contact with the starting material or product during any stage of manufacture.
Equipment used in the manufacturing process of HCT products that does not come into direct contact with the product is not a critical material.
A manufacturer is not necessarily required to perform testing on critical materials to ensure they are not contaminated with or likely to introduce bacterial or other infectious agents. Selection and evaluation of a critical material can include documents provided by the material manufacturer or suitability testing performed by the manufacturer.
Include justification of the selection and evaluation of critical material in submitted dossier.
Consider the quality of all materials that contact the product. This includes from the time of collection through to the final packaging. The clause is also designed to ensure that these materials will not adversely affect the quality, safety and efficacy of the final product. Similar clauses had been previously included in all tissue-specific TGOs. Provide a list of critical materials in the dossier for review.
To ensure the suitability of critical materials, you should review:
- material specifications provided by the material supplier OR
- testing performed by the material manufacturer, to demonstrate suitability.
Perform testing on critical materials to ensure they are suitable for use and not contaminated with, or likely to introduce, endotoxin, bacteria, or other infectious agents, if material specifications are not provided, or the documents do not provide sufficient evidence.
Justify the suitability of all critical materials used in the collection, manufacture, storage, and release of the HCT product.
Justify the suitability of all critical materials used in the collection, manufacture, storage, and release of the HCT product.
If any of the critical materials are included in the Australian Register of Therapeutic Goods (ARTG), then you:
- can cross-reference the ARTG number
- do not need to resubmit validation data for reassessment
- should include a statement justifying that the critical material is being used for the same purpose for which it is registered and that it meets sterility requirements.
Determine what requirements are applicable to critical solutions. The pharmacopoeial test for sterility is specified in the default standards under Part 3.1 of the Act. Perform a filter integrity test on the filters used to sterilise solutions. Verify that the sterilising filter is compatible with antimicrobial solutions.
Critical materials derived from human and animal sources present a unique risk for the potential transmission of viruses and Transmissible Spongiform Encephalopathy (TSE) agents. These materials must be sourced, tested (if methodology is available), and assessed as presenting a minimal risk of transmitting infectious disease agents in accordance with the requirements set out in the following documents:
- TGA guidance: Transmissible Spongiform Encephalopathies (TSE): TGA approach to minimising the risk of exposure
- European Medicine Agency (EMA) guidance: Note for Guidance on Virus Validation Studies: The design, contribution and interpretation of studies validating the inactivation and removal of viruses.
Use sterile or sterile single use equipment and containers where they are in direct contact with the starting material. If reusable equipment and containers are used, they are subject to validation. Cleaning and sterilisation processes should be monitored to minimise risk of cross-contamination.
Section 11 - Microbial contamination control strategy
11 Microbial contamination control strategy
A risk-based microbial contamination control strategy, which considers the nature of HCT materials and biologicals, must:
- be implemented to minimise intrinsic and extrinsic microbial contamination of HCT materials and biologicals; and
- specify storage, handling and transportation requirements requirements (including in relation to temperature and duration) for the HCT materials and biologicals.
This section mandates that the strategies to minimise microbial contamination must be maintained and may depend on the specific processing and uses of the biological.
It is acknowledged that some starting HCT materials are not sterile as they have inherent microflora that will not be eradicated by processing or manufacturing steps. Formulate the microbiological specifications for these finished products to specify the maximum allowable level and specify the absence of ‘specified microorganisms’, which would render the product unsuitable for use.
Section 12 - Samples for bioburden testing
12 Samples for bioburden testing
- Samples must be taken for bioburden testing.
- Samples must be:
- taken using a validated sampling technique; and
- tested using a validated test method.
Note: Subsections (1) and (2) apply in relation to all biologicals, other than human ocular tissue products to which Part 5 applies.
- This section does not apply in relation to human ocular tissue products to which Part 5 applies.
Note: Part 5 specifies bioburden testing requirements in relation to human ocular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, subsections (1) and (2) apply in relation to all other human ocular tissue products.
Use validated techniques or testing methods for bioburden sampling and testing. Validate both the sampling method and the bioburden test method, include the details of validation in the relevant section of the dossier.
Unless the entire tissue product is subject to bioburden testing, validate that a sample or portion is representative of the entire tissue. Sample from a variety of areas across the tissue during sampling validation studies and compare the bioburden test results. Take subsequent routine bioburden samples from any area which represents the ‘worst case’ in terms of bioburden.
Section 13 – Bioburden testing requirements
13 Bioburden testing requirements
- Written specifications for HCT materials and biologicals must include specified microorganisms determined on the basis of a risk assessment.
- Samples must demonstrate that HCT materials are free from contamination with specified microorganisms.
- Samples must demonstrate that biologicals are:
- free from microbial contamination; or
- if, on the basis of a risk assessment, it is not necessary for the biological to be free from microbial contamination—free from contamination with specified microorganisms.
- This section does not apply in relation to:
- human cardiovascular tissue products to which Part 4 applies; and
- human ocular tissue products to which Part 5 applies.
Note 1: Part 4 provides that human cardiovascular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only, and human cardiovascular tissue used in the manufacture of those products, must be free from microbial contamination. However, this section applies in relation to all other human cardiovascular tissue products.
Note 2: Part 5 specifies bioburden testing requirements in relation to human ocular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, this section applies in relation to all other human ocular tissue products.
- Subsection (3) does not apply in relation to:
- human musculoskeletal tissue products to which Part 3 applies; and
- human skin products to which Part 6 applies.
Note 1: Part 3 provides that human musculoskeletal tissue products that are subjected to only minimal manipulation must be free from microbial contamination, and human musculoskeletal tissue used in the manufacture of those products must be free from contamination with specified microorganisms. However, subsection (3) applies in relation to all other human musculoskeletal tissue products.
Note 2: Part 6 provides that human skin products that are subjected to only minimal manipulation and human skin used in the manufacture of those products must be free from contamination with specified microorganisms. However, subsection (3) applies in relation to all other human skin products.
This section applies to any HCT material (collected or during processing) and the finished product (biological).
Use validated methods to sample and test the biological to show the absence of microorganisms or specified microorganisms. Provide the details in the relevant section of the dossier.
Use a risk-based approach to develop the list of specified microorganisms of clinical significance for the product, which, if present, require rejection of the tissue for clinical use. Include consideration of the category of HCT, the method of processing, and the nature and type of microorganisms which might be present. Include the list of microorganisms in the appropriate dossier section.
The Council of Europe’s 4th Edition of the Guide to the quality and safety of tissues and cells for human application (2019) provides guidance around microbial contaminants that should result in tissue discard, although these lists are not exhaustive, see:
- Chapter 18, Table 18.2 for amniotic membrane
- Chapter 19, Table 19.2 for skin
- Chapter 20, Table 20.3 for cardiovascular tissue
- Chapter 21, Table 21.2 for musculoskeletal tissue.
Sponsors should provide their own documented list of specified microorganisms and justification.
Bioburden challenge method
Pharmacopoeial and ISO standards for bioburden test methods include a requirement to validate the methods. This involves challenging the material or product with low numbers (< 100 CFU) of reference challenge microorganisms and recovering these organisms within the shortest test incubation time.
Given that antimicrobial agents could be present in the starting materials and/or the end HCT product, it is necessary to attempt to neutralise these agents to optimise the recovery of the challenge microorganisms, and any product contaminants. Pharmacopoeial and ISO methods mandate this step under ‘suitability of test method’ or ‘method validation’.
Antimicrobial activity can often be removed by filtration, dilution and/or chemical inactivation using a suitable neutralising agent.
Tissue banks should attempt to identify antimicrobial agents used to treat donors and agents during processing to assist them identify suitable neutralising agents. If antimicrobial properties cannot be neutralised, pharmacopoeias permit the product to be tested under the set of conditions established as optimal for recovery. Justify this approach and provide details for assessment or audit by TGA.
Microbial testing outcomes
HCT material which demonstrates microbial contamination provide two possible outcomes:
- the HCT material or biological must be deemed unsuitable for therapeutic use OR
- provided that specified microbes are excluded, the HCT material or biological may be processed or released.
When a bioburden reduction treatment renders a biological ‘free from microbial contamination’, that biological is not regarded as sterile unless the process was validated as a sterilisation process and the product passes a pharmacopoeial sterility test.
The microbial contamination sections of:
- the pharmacopoeial default standards
- ISO/DTS 22456: Sterilization of healthcare products - Microbiological Methods - Guidance on conducting bioburden determinations and tests of sterility for tissue-based products
- ISO 14160: Sterilization of health care products - Liquid chemical sterilizing agents for single-use medical devices utilizing animal tissues and their derivatives - Requirements for characterization, development, validation, and routine control of a sterilization process for medical devices.
provide useful guidance on suitable bioburden test methods and their validation to demonstrate neutralisation/inactivation of antimicrobial substances. ISO 11737-1 specifically describes steps to establish the recovery efficiency and correction factor(s) that are to be applied when testing bioburden on/within solid and semi-solid starting materials (for example, bone, tendon).
Section 14 - Sterilisation
14 Sterilisation
The sterilisation process for a biological that is terminally sterilised must be validated to ensure a maximal sterility assurance level of 10-6
If the product is to be terminally sterilised, perform validation and routine control of sterilisation processes in accordance with the relevant parts of international sterilisation standards, for example:
- ISO 11135 (ethylene oxide)
- ISO 14937 (general methods)
- ISO 17665 (moist heat)
- ISO 11137 (radiation)
- ISO 14160 (chemical sterilisation)
- ISO 20857 (dry heat).
These are examples of sterilisation methods that could be used for terminal sterilisation of biologicals. There may be other methods that are suitable for use in terminal sterilisation of biological products, provided that they can be validated and routinely controlled to produce the specified sterility assurance level (SAL).
Such methods would likely be covered by the validation and routine control procedures specified in ISO 14937.
Some of these standards are relevant to the sterilisation of containers and ancillary materials used during tissue processing. Any of the ISO standards listed above provide guidance for validating the relevant sterilisation process for containers. Sterile containers must be used, these can be pre-purchased as sterile.
Section 15 - Collection from deceased donors
15 Collection from deceased donors
- HCT materials must be collected from a deceased donor as soon as possible after asystole, and collection must be completed:
- if the body has been refrigerated below 10°C within 12 hours of asystole—not later than 24 hours after asystole; or
- if paragraph (a) does not apply—not later than 15 hours after asystole.
- This section does not apply in relation to:
- human musculoskeletal tissue products to which Part 3 applies; and
- human ocular tissue products to which Part 5 applies; and
- human amnion products to which Part 7 applies.
Note 1: Part 3 provides requirements in relation to the collection of human musculoskeletal tissue used in the manufacture of human musculoskeletal tissue products that are subjected to only minimal manipulation. However, this section applies in relation to all other human musculoskeletal tissue products.
Note 2: Part 5 provides requirements in relation to the collection of human ocular tissue used in the manufacture of human ocular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, this section applies in relation to all other human ocular tissue products.
Note 3: Part 7 provides requirements in relation to the collection of human amnion used in the manufacture of human amnion products that are subjected to only minimal manipulation. However, this section applies in relation to all other human amnion products.
When collecting HCT material from deceased donors, the time between asystole and the commencement of the collection process must be as small as possible to minimise the proliferation of microbial contaminants.
The timeframes detailed in this clause are the default collection timeframes. Specific tissue types may have more stringent or more relaxed collection criteria defined in Parts 3-7 of TGO 109.
If collection timeframes are longer than those defined above, validate the quality of the starting material to demonstrate that extending the time before collection does not compromise the quality, safety or efficacy of the HCT product manufactured from the starting material.
Section 16 - Storage and transportation: HCT materials
16 Storage and transportation
HCT materials
- Immediately following collection and prior to processing, HCT materials must be:
- stored as follows:
- at less than 10°C for not longer than 72 hours; or
- as otherwise validated by the manufacturer to prevent microbial proliferation and to ensure the quality, safety and efficacy of the biological manufactured using the HCT materials; and
- transported in a manner that ensures the relevant storage conditions specified in paragraph (a) are maintained during transportation.
- Subsection (1) does not apply in relation to:
- human cardiovascular tissue products to which Part 4 applies; and
- human skin products to which Part 6 applies
- human amnion products to which Part 7 applies.
Note 1: Part 4 provides storage and transportation requirements in relation to cardiovascular tissue used in the manufacture of cardiovascular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, subsection (1) applies in relation to all other human cardiovascular tissue products.
Note 2: Part 6 provides storage and transportation requirements in relation to human skin used in the manufacture of human skin products that are subjected to only minimal manipulation. However, subsection (1) applies in relation to all other human skin products.
Note 3: Part 7 provides storage and transportation requirements in relation to human amnion used in the manufacture of human amnion products that are subjected to only minimal manipulation. However, subsection (1) applies in relation to all other human amnion products.
- HCT materials that are collected and transported for use in the manufacture of a biological must be packaged using aseptic technique and with at least one moisture impermeable barrier.
This section provides a default temperature and time between the collection of starting material and commencement of processing. Validate timeframes outside of these parameters to demonstrate that the starting material (HCT material) quality is not affected, especially with respect to proliferation of microbial bioburden.
Unless alternative timeframes have been defined in Parts 3-7 of TGO 109, the default collection to processing timeframe should be less than 72 hours, with the starting material stored below 10°C.
Validate proposed pre-processing storage conditions if the collected starting material may be stored before the commencement of processing.
Provide data to demonstrate that the quality and safety of the biological will not be adversely affected if longer pre-processing storage or higher storage temperatures are used. Validation data should demonstrate that the HCT product manufactured from stored starting material can meet all product release specifications. Use this data to inform process control points that provide limits to pre-processing storage times.
Section 16 – Storage and transportation: biologicals
16 Storage and transportation
Biologicals
- Biologicals must be stored and transported under conditions that are validated, including justified time and temperature specifications, to ensure the quality and safety of the biologicals.
- Subsection (4) does not apply in relation to:
- human musculoskeletal tissue products to which Part 3 applies; and
- human cardiovascular tissue products to which Part 4 applies; and
- human ocular tissue products to which Part 5 applies; and
- human skin products to which Part 6 applies; and
- human amnion products to which Part 7 applies.
Note 1: Part 3 provides storage and transportation requirements in relation to human musculoskeletal tissue products that are subjected to only minimal manipulation. However, subsection (4) applies in relation to all other human musculoskeletal tissue products.
Note 2: Part 4 provides storage and transportation requirements in relation to human cardiovascular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, subsection (4) applies in relation to all other human cardiovascular tissue products.
Note 3: Part 5 provides storage and transportation requirements in relation to human ocular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, subsection (4) applies in relation to all other human ocular tissue products.
Note 4: Part 6 provides storage and transportation requirements in relation to human skin products that are subjected to only minimal manipulation. However subsection (4) applies in relation to all other human skin products.
Note 5: Part 7 provides storage and transportation requirements in relation to human amnion products that are subjected to only minimal manipulation. However, subsection (4) applies in relation to all other human amnion products.
- A biological that is returned to its manufacturer cannot be released for supply unless:
- the biological was at all times stored and transported in accordance with applicable storage and transportation requirements in relation to the biological; and
- the labelling and packaging of the biological has not been compromised.
Validate the storage conditions of the HCT product (the biological) to ensure that the quality is not compromised.
The purpose of this clause is to ensure that the manufacturer has validated the shelf life and storage conditions of the final product. Tissue-specific parts provide ‘default’ shelf life based on international best practise for different tissue types. This clause captures the same requirement for all other biological products.
Provide data demonstrating that the shelf life, as defined by proposed storage conditions and times, maintains the quality and safety of the HCT product. Measure critical quality parameters for at least three batches of product at regular intervals to confirm that product quality is maintained. Demonstrate integrity and suitability of the container systems for the proposed storage conditions and shelf life.
A biological that has undergone a deviation to the recommended storage conditions may only be supplied if TGA has assessed the data to support the release. This data may be submitted in the original application, or as a variation to the proposed shelf life. Validate any deviations from proposed storage conditions (for example, temperature excursions, thermal cycling) using at least three batches of the product.
The following guidance (for prescription medicines) may be of use in evaluating the shelf life of a biological product and for the design of stability studies for your HCT product:
- TGA guidance: Stability testing for prescription medicines
- EMA guidance: Quality of biotechnological products: Stability Testing of Biotechnological/Biological Products
Ensure procedures are in place to actively monitor, or validate, the transportation of the allograft after release to ensure that storage conditions are maintained.
Allowance has been made for manufacturers to justify and set their own transport and storage temperature requirements after processing (unless defined in Parts 3-7 of TGO 109). Specify the conditions of product transport and storage to ensure maintenance of product quality including minimisation of microbial contamination. Conduct active monitoring of product temperature and conditions following release of product. Alternatively, validate the transportation process, and limit process to validated temperatures and timeframes.
If the manufacturer intends to accept return of a biological, they must have a documented system in place to ensure that the quality, safety, and efficacy of the biological has not been compromised between release and return. This includes procedures to ensure that the product has been stored at the required temperatures at all times following release or has only been exposed to temperature excursions that have been demonstrated (through validation studies) to not compromise product quality. Provide these procedures in the dossier for TGA review.
Section 17 - Containers of biologicals
17 Containers of biologicals
- A biological must be sealed within a sterile container and must be at least double packaged so as to:
- prevent ingress or egress of all materials; and
- ensure that any breach of integrity of the container and packaging is evident.
- This section does not apply in relation to human ocular tissue products to which Part 5 applies.
Note: Part 5 provides requirements in relation to containers of human ocular tissue products that are subjected to only minimal manipulation and are manufactured for allogeneic use only. However, this section applies in relation to all other human ocular tissue products.
Ensure sterile packaging is compatible with the product and if terminally sterilised, with the method of sterilisation. For example, the packaging should allow the ingress of the sterilising agent and where applicable, its egress. It is permissible to sterilise the container along with the HCT.
The use of new technology (for example, supercritical CO2) would be considered on a case-by-case basis, based on validation data which demonstrates that there is a maximal sterility assurance level (SAL) of 10-6 for a terminal sterilisation process. Guidance on novel sterilisation method validation can be found in ISO 14937.
If the tissue is aseptically manufactured, then it should be transferred into a sterile container and validated as an aseptic process (refer to ISO 13408 and ISO 18362).
Part 3 – Standard for human musculoskeletal tissue products
Section 18 – What this Part is about
18 What this Part is about
This Part specifies requirements that must be met in the manufacture of human musculoskeletal tissue products that have been subjected to only minimal manipulation, including requirements relating to human musculoskeletal tissue that is collected for use in the manufacture of those products.
Unless otherwise specified, a biological to which this Part applies must meet the general requirements in Part 2 in addition to the specific requirements in this Part.
Part 3 applies to human musculoskeletal tissue, including:
- muscle
- ligament
- tendon
- fascia lata
- bone
- cartilage.
Section 19 - Application of this Part
19 Application of this part
This part applies in relation to human musculoskeletal tissue products that have been subjected to only minimal manipulation.
Minimal manipulation is defined by TGA here:
Section 20 - Collection from deceased donors
20 Collection from deceased donors
- The collection of human musculoskeletal tissue from a deceased donor must commence as soon as possible after asystole and:
- if the body has been refrigerated below 10°C within 12 hours after asystole—not later than 24 hours after asystole; or
- if paragraph (a) does not apply—not later than 15 hours after asystole.
- The collection of human musculoskeletal tissue from a deceased donor must be completed within 36 hours after asystole.
Commence collection of musculoskeletal tissue as soon as possible after asystole with the maximum allowable time for completion of the collection being 36 hours.
The time post-asystole at which the collection of musculoskeletal tissue must commence is dependent on the temperature at which the body has been stored: if it has been refrigerated below 10°C within 12 hours of asystole, then collection must commence within 24 hours; otherwise, collection must commence within 15 hours.
The application of antiseptic solution to decontaminate the donor’s skin (skin preparation) is generally accepted as the commencement of the collection process as defined in the American Association for Tissue Banks (AATB) guidance.
Section 21 - Bioburden testing
21 Bioburden testing
Tissue not subjected to processing
- Human musculoskeletal tissue that is not subjected to processing prior to packaging must be sampled at the time of collection for bioburden testing.
- Human musculoskeletal tissue sampled in accordance with subsection (1) must be tested for bioburden and:
- demonstrate no microbial contamination; or
- be rejected for therapeutic use if microbial contamination is demonstrated; or
- be subjected to further processing in accordance with subsection (3) if microbial contamination is demonstrated with microorganisms other than specified microorganisms.
Tissue subjected to processing- Human musculoskeletal tissue that is subjected to processing prior to packaging (including, for example, a bioburden reduction process) must be:
- sampled for bioburden testing at the time of collection or prior to the processing to exclude tissue contaminated with specified microorganisms; and
- either:
- sampled for bioburden testing after the processing and must demonstrate no microbial contamination; or
- subjected to a bioburden reduction process that has been validated to render the tissue free from any microbial contamination.
- Human musculoskeletal tissue that demonstrates:
- contamination with specified microorganisms when tested in accordance with paragraph 21(3)(a); or
- any microbial contamination when tested in accordance with subparagraph 21(3)(b)(i);
must not be used for therapeutic use.
For musculoskeletal tissue that is not to be processed after collection, sample the tissue at the time of collection for bioburden testing. Sample should be shown to be free of microbial contamination. Use validated sampling method and bioburden test method, include the details of validation in the relevant section of the dossier.
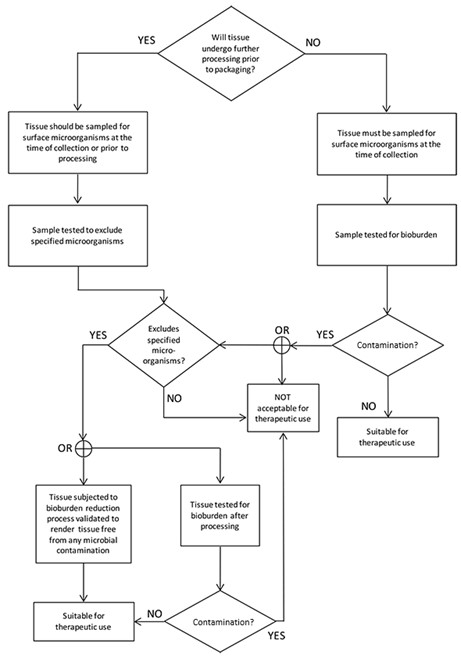
The flowchart in Figure 1 shows how the requirements identified in this section establish the suitability of the collected musculoskeletal tissue for therapeutic use.
Figure 1 – Flowchart for musculoskeletal tissue
{kind=link}
Flow chart depicting tissue processing decision tree.
The chart starts with the question 'Will tissue undergo further processing prior to packaging?'.
If YES, tissue is sampled for surface microorganisms at time of collection or prior to processing, then tested to exclude specified microorganisms. If it excludes them, tissue is subjected to bioburden reduction process validated to result in sterility.
If NO to initial question or if specified microorganisms not excluded, tissue must be sampled for surface microorganisms at time of collection, then tested for bioburden.
Both paths lead to checking for contamination.
If contaminated, it's not acceptable for therapeutic use.
If not contaminated, it's suitable for therapeutic use.
The process ensures tissue is either sterile or has acceptable bioburden levels before being deemed suitable for therapeutic use
There are two possible outcomes for collected and sampled musculoskeletal tissue which demonstrates microbial contamination. The tissue must be either:
- deemed unsuitable for therapeutic use
- provided that specified microbes are excluded (see Part 2), the tissue can be subject to further processing to render it free from microbial contamination (as detailed in Part 3) Sample tissues at the time of collection for bioburden. When contaminated with specified microorganisms (see Part 2), the tissue should be excluded from therapeutic use.
If not contaminated with specified microorganisms, the tissue should be either:
- sampled post-bioburden reduction and demonstrate no microbial contamination
- subjected to a bioburden reduction process that has been validated to render the tissue free from microbial contamination.
Both the sampling method and the bioburden test method must be validated, the details of which should be included in the relevant section of the dossier. When a bioburden reduction treatment renders a tissue ‘free from microbial contamination’, the tissue is not regarded as sterile unless the process was validated as a sterilisation process and the product passes a pharmacopoeial sterility test.
For guidance on the validation of bioburden sampling method, bioburden challenge method, and validation of bioburden test method, see Part 2 Standard for all biologicals.
Do not use any musculoskeletal tissue for therapeutic use that demonstrates contamination of specified microorganisms post-collection. Do not use tissues for therapeutic use where any microbial contamination is detected post-processing.
Section 22 - Demineralisation products
22 Demineralised products
The residual calcium of human musculoskeletal tissue products that are demineralised must not exceed 8% w/w.
For a bone product to be classified as demineralised, it must have a reduction in calcium levels. This specification is adopted from the American Association for Tissue Banks (AATB) guidance which mandates residual calcium levels for demineralised bone products.
Unless bone is treated by a validated process to reduce minerals, test representative samples of each lot for residual calcium using a validated standard method. Residual calcium content for bone labelled as ‘demineralised’ shall not exceed 8% w/w according to a standard method. For bone that has been subjected to a demineralisation process with a residual calcium content target that exceeds 8% w/w when tested, the tissue must not be labelled as ‘demineralised’. Labelled as ‘partially demineralised’ to describe the extent of demineralisation.
Some demineralised bone may claim osteoinductive properties. Do not make an osteoinductive claim unless the osteoinductive potential of the product has been demonstrated by a validated test method.
Section 23 – Freeze-dried products
23 Freeze-dried products
The residual moisture content of human musculoskeletal tissue products that are freeze-dried must not exceed 6% w/w.
Freeze-drying is also known as lyophilisation and involves decreasing the water content of tissue, usually under vacuum through sublimation.
For a product to be labelled as ‘freeze-dried’, it must have a residual moisture content of ≤6% w/w. Freeze-dried products can be stored at room temperature.
This specification has been adopted from the Council of Europe’s 4th Edition of the Guide to the quality and safety of tissues and cells for human application (2019) and current dossiers for freeze-dried products.
Section 24 – Storage and transportation
24 Storage and transportation
- Human musculoskeletal tissue products must be: stored as follows:
- at minus 20°C to minus 40°C for not more than 6 months after the collection of the human musculoskeletal tissue; or
- frozen or cryopreserved at less than minus 40°C for not more than 5 years after the collection of the human musculoskeletal tissue; or
- if the products are freeze-dried—at room temperature for not more than 5 years after the collection of the human musculoskeletal tissue; or
- in accordance with conditions that are validated on the basis of validated data or documented evidence from relevant scientific literature, including justified time and temperature specifications, to ensure the quality, safety and efficacy of the products; and
- transported in a manner that ensures the relevant storage conditions specified in paragraph (a) are maintained during transportation.
Because the storage conditions for musculoskeletal tissue are stated in TGO 109, validation of these storage conditions is not required. However, validation of shelf life for packaging materials still needs to be performed. Specify the conditions of product storage to ensure maintenance of product quality, including the minimisation of microbial contamination.
Perform validation studies if you wish to have an extended shelf life.
Please note that in addition to TGO 109 requirements, all relevant requirements of TGO 107 (labelling of biologicals) apply to musculoskeletal tissue. If further clarification is required, please contact TGA’s Biological Science Section (BSS).
Part 4 – Standard for human cardiovascular tissue products
Section 25 – What this Part is about
25 What this Part is about
This Part specifies requirements that must be met in the manufacture of human cardiovascular tissue products that have been subjected to only minimal manipulation and are manufactured for allogeneic use only, including requirements relating to human cardiovascular tissue that is collected for use in the manufacture of those products.
Unless otherwise specified, a biological to which this Part applies must meet the general requirements in Part 2 in addition to the specific requirements in this Part.
Part 4 applies to human cardiovascular tissue including:
- Aortic
- Pulmonary
- mitral and tricuspid heart valves, or any part of such valves
- vascular tissue.
Vascular tissue may include conduit or greater vessel graft, peripheral vascular tissue graft, pericardial graft.
Section 26 – Application of this Part
26 Application of this Part
This Part applies in relation to human cardiovascular tissue products that:
- have been subjected to only minimal manipulation; and
- are manufactured for allogeneic use only.
Minimal manipulation is defined by TGA here:
- TGA guidance: Method of preparation: Interpretation of minimal manipulation
Allogeneic use, in relation to an HCT product, means administration to, or application in the treatment of, a person other than the person from whom the HCT materials used in the manufacture of the product were collected.
Section 27 – Tissue not subjected to a bioburden reduction process
27 Tissue not subjected to a bioburden reduction process
- Human cardiovascular tissue that is not subjected to a bioburden reduction process must be collected, subjected to any processing prior to packaging, sampled for bioburden testing, packaged in an operating theatre, transported to a manufacturing site, and cryopreserved.
- The cryopreservation of human cardiovascular tissue mentioned in subsection (1) must commence:
- in the case of a deceased donor—within 48 hours of asystole; or
- in the case of a living donor—within 48 hours of collection.
- Human cardiovascular tissue that is sampled in accordance with subsection (1) must be tested for bioburden and, following that testing, must:
- demonstrate no microbial contamination; or
- be rejected for therapeutic use if microbial contamination is demonstrated.
When a bioburden reduction process will not be applied to cardiovascular tissue, the time from collection (from a living donor or asystole of a deceased donor), to the commencement of cryopreservation of the packaged allograft tissue, must be less than 48 hours.
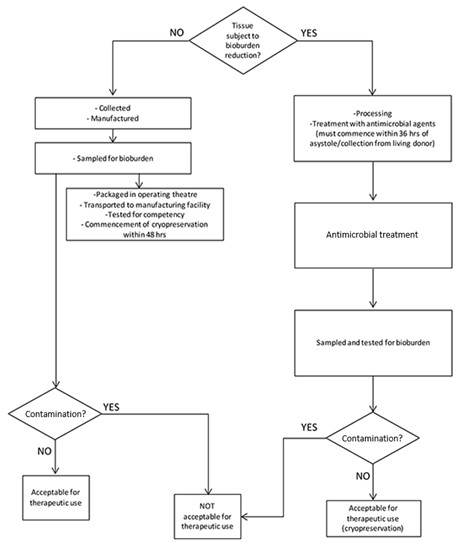
The flowchart in Figure 2 shows how the requirements identified in this section and Part 4 establish the suitability of the collected cardiovascular tissue for therapeutic use.
Figure 2 – Flowchart for cardiovascular tissue
{kind=link}
Flow chart showing tissue bioburden reduction process.
It begins by asking if tissue is subject to bioburden reduction.
If NO, tissue is collected, manufactured, sampled for bioburden, transported to manufacturing facility, tested for sterility, and cryopreserved within 48 hrs.
If YES, tissue undergoes treatment with antimicrobial agents within 3 hrs of asystole/collection from living donor, followed by antimicrobial treatment.
Both paths lead to sampling and testing for bioburden, then checking for contamination.
If no contamination is found, tissue is acceptable for therapeutic use. If contamination is present, it's not acceptable for therapeutic use.
The chart ensures proper handling and testing of tissue to determine its suitability for therapeutic applications
For guidance on the validation of bioburden sampling method and validation of bioburden test method, see Part 2 – Standard for all biologicals.
For guidance on the bioburden challenge method, see Part 2 – Standard for all biologicals.
If bioburden test results fall outside the product specifications, reject the tissue product.
Section 28 - Tissue subjected to a bioburden reduction process
28 Tissue subjected to a bioburden reduction process
- Human cardiovascular tissue that is subjected to a bioburden reduction process must be processed and treated with antimicrobial agents in accordance with subsections (2) and (3).
- The processing and treatment with antimicrobial agents must commence:
- in the case of a deceased donor—within 36 hours of asystole; or
- in the case of a living donor—within 36 hours of collection.
- The human cardiovascular tissue must be exposed, between dissection of the tissue from its surrounding tissue and the time of its cryopreservation, to conditions of antimicrobial treatment at:
- 34°C to 39°C for 6 to 12 hours; or
- 2°C to 8°C for 18 to 24 hours.
- Following the bioburden reduction process and before the addition of cryopreservative, human cardiovascular tissue must be sampled, and tested for bioburden and, following testing, must:
- demonstrate no microbial contamination; or
- be rejected for therapeutic use if microbial contamination is demonstrated.
- Human cardiovascular tissue that is subjected to a bioburden reduction process must be cryopreserved:
- within 72 hours of asystole; or
- within a timeframe that is validated by the manufacturer to prevent microbial proliferation and to ensure the quality, safety and efficacy of the human cardiovascular product manufactured using the human tissue.
When a bioburden reduction process will be applied to cardiovascular tissue, the time from collection from a donor to the treatment of antimicrobial agents must commence within 36 hours.
The flowchart in Figure 2 shows how the requirements identified in this section and Part 4 establishes the suitability of the collected cardiovascular tissue for therapeutic use.
For guidance on the validation of bioburden sampling method and validation of bioburden test method, see Part 2 – Standard for all biologicals.
The entire manufacturing process (completion of cryopreservation) must be completed within 72 hours of asystole or validated. This is consistent with the Council of Europe’s 4th Edition of the Guide to the quality and safety of tissues and cells for human application (2019) recommendation. Manufacturing timeframes longer than this should be justified and validated to ensure that the quality and safety of the allograft tissue is not adversely affected.
Section 29 - Heart valves
29 Heart valves
Human cardiovascular tissue that is a heart valve must be a competent valve prior to cryopreservation.
Detail the tests for heart valve competency and justify the choice of method(s).
Section 30 – Storage and transportation
30 Storage and transportation
Human cardiovascular tissue
- Immediately following collection and prior to processing in accordance with either section 27 or 28, human cardiovascular tissue must be:
- stored as follows:
- temperature between 0°C to 10°C; or
- as otherwise validated by the manufacturer to prevent microbial proliferation and to ensure the quality, safety and efficacy of the human cardiovascular tissue product manufactured using the tissue; and
- transported in a manner that ensures the relevant storage conditions specified in paragraph (a) are maintained during transportation.
Human cardiovascular tissue products
- Human cardiovascular tissue products that are cryopreserved must be stored as follows:
- at or below minus 100°C for not more than 5 years after collection of the human cardiovascular tissue; or
- in accordance with conditions that are validated on the basis of validated data or documented evidence from relevant scientific literature, including justified time and temperature specifications, to ensure the quality, safety and efficacy of the products.
- Human cardiovascular tissue products that are cryopreserved must be transported in a manner that ensures the relevant storage conditions specified in subsection (2) are maintained during transportation.
This section provides a default temperature for cardiovascular tissue storage between the collection of starting material and commencement of processing. Validate temperatures outside of these parameters to demonstrate that the starting material quality is not affected, especially with respect to proliferation of microbial bioburden.
If longer pre-processing storage or higher storage temperatures are used, provide data to demonstrate that the quality and safety of the final HCT product will not be adversely affected. Validation data should demonstrate that the HCT product manufactured from stored starting material can meet all product release specifications. Use this data to inform process control points that provide limits to pre-processing storage times.
Validate the storage conditions of the HCT product during the manufacturing process to ensure that the quality of the final product is not compromised. Process validation studies should: determine the length of time and temperature of the processing takes place (defining normal operational limits of the manufacturing process), and validate in-process hold times for product intermediates (including in-process freeze/thaw cycles).
Validation of cryopreserved cardiovascular tissue conditions is not required as they are stated in this order (that is, at or below minus 100°C for no more than five years). Perform full validation if storage conditions for non-cryopreserved cardiovascular tissue are implemented.
Please note that in addition to TGO 109 requirements, all relevant requirements of TGO 107 (labelling of biologicals) apply to cardiovascular tissue. If further clarification is required, please contact TGA’s Biological Science Section (BSS).
Part 5 – Standard for human ocular tissue products
Section 31 – What this Part is about
31 What this Part is about
This Part specifies requirements that must be met in the manufacture of human ocular tissue products that have been subjected to only minimal manipulation and are manufactured for allogeneic use only, including requirements relating to human ocular tissue that is collected for use in the manufacture of those products.
Unless otherwise specified, a biological to which this Part applies must meet the general requirements in Part 2 in addition to the specific requirements in this Part.
Part 5 applies to biologicals that are human ocular tissue, including:
- eye globe
- cornea
- sclera.
Section 32 – Application of this Part
32 Application of this Part
This Part applies in relation to human ocular tissue products that:
- have been subjected to only minimal manipulation; and
- are manufactured for allogeneic use only.
Minimal manipulation is defined by TGA here:
Allogeneic use, in relation to an HCT product, means administration to, or application in the treatment of, a person other than the person from whom the HCT materials used in the manufacture of the product were collected.
Section 33 – Collection
33 Collection
- Collection of human ocular tissue from a deceased donor must be completed not later than 48 hours after asystole, and the time intervals between asystole, enucleation, preservation and corneal excision must be documented.
- A human ocular tissue product that is manufactured using human ocular tissue collected between 24 and 48 hours after asystole must not be released for supply unless:
- a medical practitioner acting for, or on behalf of, the manufacturer or the sponsor has evaluated the quality, safety and efficacy of the product; and
- the medical practitioner who is responsible for the administration to, or application in the treatment of, the recipient of the product has been notified that the collection occurred between 24 and 48 hours after asystole; and
- the medical practitioners mentioned in paragraphs (a) and (b) are registered in a State or internal territory.
Where ocular tissue is removed from the stated storage conditions to allow quality determination, define the process and provide sound scientific justification as to why the quality and safety is not compromised. An example may include warming to room temperature to optimise microscopic evaluation and/or endothelial cell counting.
Section 34 – Storage and transportation
34 Storage and transportation
Human ocular tissue products must be stored as follows:
- in the case of an eye globe—in a moist chamber system at 0°C to 10°C for not more than 48 hours after collection of the eye globe; or
- in the case of excised cornea—either:
- in a corneal storage medium at 0°C to 10°C for not more than 14 days after collection of the cornea; or
- in a storage medium at 28°C to 37°C for not more than 30 days after collection of the cornea; or
- in a cryopreservation medium between minus 75°C to minus 196°C for not more than 2 years after the collection of the cornea; or
- in the case of sclera—maintained in at least 75% v/v ethanol solution for not more than 1 year after collection of the sclera; or
- in accordance with conditions that are validated on the basis of validated data or documented evidence from relevant scientific literature, including justified time and temperature specifications, to ensure the quality, safety and efficacy of the products.
Sponsors may be granted a shelf life for ocular products as outlined above.
Perform validation of shelf life for packaging materials. Specify the conditions of product storage to ensure maintenance of product quality, including the minimisation of microbial contamination.
Section 35 – Excised cornea – testing of storage medium etc.
35 Excised cornea – testing of storage medium etc.
- Where excised cornea is stored in accordance with subparagraph 34(b)(ii), then:
- the storage medium must be tested for microbial contamination using a validated test method prior to transfer of the tissue to a transport medium; and
- subsequent exposure to the transport medium at a temperature validated to maintain tissue quality must not exceed 5 days.
- If testing of the storage medium in accordance with paragraph (1)(a) demonstrates evidence of microbial contamination, then:
- where the excised cornea has not been released for supply—the excised cornea must be rejected for therapeutic use; or
- where the excised cornea has been released for supply—the results of the microbial tests must be reported to the medical practitioner who is responsible for the administration to, or application in the treatment of, the recipient of the product in accordance with the documented procedures of the manufacturer.
This section only applies to excised cornea maintained in storage medium at 28-37°C for no more than 30 days.
Use sterile storage and transport solutions in processing ocular tissue. Test storage medium that is intended to be sterile using the harmonised test for sterility by the filtration method specified in the default standard pharmacopoeias.
Storage medium must pass the test. Demonstrate neutralisation of antimicrobial agents that might interfere with the growth of contaminants in validation of the test method described as the ‘suitability of test’.
Alternatively, validate the filtration method as described in the microbial contamination chapters of the pharmacopoeias to demonstrate the absence of growth in solutions not required to be sterile.
For guidance on the validation of bioburden sampling method and validation of bioburden test method, see Part 2 – Standard for all biologicals.
For guidance on the bioburden challenge method, see Part 2 – Standard for all biologicals.
Section 36 – Containers
36 Containers
A human ocular tissue product must be sealed within a sterile container and must be packaged so as to:
- prevent ingress or egress of all materials; and
- ensure that any breach of integrity of the container and packaging is evident.
The sterile packaging should be compatible with the product and if terminally sterilised, with the method of sterilisation, for example, the packaging should allow the ingress of the sterilising agent and where applicable, its egress. It is permissible to sterilise the container along with the HCT.
The use of new technology (for example, supercritical CO2) would be considered based on evidence of validation to demonstrate that there is a maximal sterility assurance level (SAL) of 10-6 for a terminal sterilisation process. Guidance on novel sterilisation method validation can be found in ISO 14937.
Transfer aseptically manufactured tissue into a sterile container. Validate as an aseptic process (refer to ISO 13408 and ISO 18362).
Section 37 – Examination and evaluation
37 Examination and evaluation
Examination and evaluation of human ocular tissue must be in accordance with the requirements set out in section 10 of the EBAANZ Standards for Eye Donation and Eye Tissue Banking.
Evaluation and examination methods for ocular tissue, including cornea endothelial cells, are specified in The Eye Bank Association of Australia and New Zealand (EBAANZ) Medical and Quality Standards for Eye Donation and Eye Tissue Banking (Chapter 10, Edition 2, April 2009).
Where ocular tissue is removed from the stated storage conditions to allow quality determination (for example, warmed to room temperature to optimise microscopic evaluation and/or endothelial cell counting), define the process and provide sound scientific justification as to why the quality and safety of the tissue is not compromised.
Please note that in addition to TGO 109 requirements, all relevant requirements of TGO 107 (labelling of biologicals) apply to human ocular tissue. If further clarification is required, please contact TGA’s Biological Science Section (BSS).
Part 6 – Standard for human skin products
Section 38 – What this Part is about
38 What this Part is about
This Part specifies requirements that must be met in the manufacture of human skin products that have been subjected to only minimal manipulation, including requirements relating to human skin that is collected for use in the manufacture of those products.
Unless otherwise specified, a biological to which this Part applies must meet the general requirements in Part 2 in addition to the specific requirements in this Part.
Part 6 applies to biologicals that are human skin.
Section 39 – Application of this
Part 39 Application of this Part
This Part applies in relation to human skin products that have been subjected to only minimal manipulation.
Minimal manipulation is defined by TGA here:
Section 40 – Processing etc.
40 Processing etc.
- Human skin must be sampled for bioburden testing following collection and prior to being packaged.
- Processing of human skin must commence:
- within 24 hours of collection; or
- within a longer timeframe that is validated by the manufacturer to prevent microbial proliferation and to ensure the quality, safety and efficacy of the human skin product manufactured using the human skin.
This section specifies the bioburden sampling and packaging requirements for collected skin. Validate both sampling and packaging processes to demonstrate compliance with TGO 109.
Unless the entire tissue product is subject to bioburden testing, validate that a sample or portion is representative of the entire tissue. Sample from a variety of areas across the tissue during sampling validation studies and compare the bioburden test results. Take subsequent routine bioburden samples from any area which represents the ‘worst case’ in terms of bioburden.
Commence tissue processing within 24 hours of procurement. Before processing, keep the recovered skin in a temperature-controlled refrigerator at 2-8°C, without interruption throughout the refrigeration process. It is recommended to change cell nutrient medium used for viable grafts shortly after receipt of skin grafts, or that you validate the medium for 24 hours storage (that is, adequate buffering capacity). Perform all the manipulations where the transport containers are going to be exposed and the media changed in a controlled-air environment.
It should be noted that ‘commencement of processing’ is a broad term that may include the commencement of the bioburden reduction step in processing (addition of antimicrobial compounds) as required by Part 6.
Section 41 – Bioburden testing
41 Bioburden testing
- Human skin sampled in accordance with subsection 40(1) must be tested for bioburden and, following that testing, must:
- demonstrate no microbial contamination with specified microorganisms; or
- be rejected for therapeutic use if microbial contamination with specified microorganisms is demonstrated.
- If bioburden testing in accordance with subsection (1) demonstrates microbial contamination with microorganisms other than specified microorganisms, then the results of the testing must be reported by the manufacturer of the human skin product to the medical practitioner who is responsible for the administration to, or application in the treatment of, the recipient of the product.
Use validated bioburden sampling and testing techniques or methods. Provide details of sampling method and bioburden test method validation in the relevant section of the dossier.
For guidance on the validation of bioburden sampling method and validation of bioburden test method, see Part 2 – Standard for all biologicals.
ISO 14160 Annex A.5.3 provides guidance on performing bioburden tests on animal tissues.
Develop a documented list with all relevant microorganisms that, if present, would result in a rejection of the product.
It is acknowledged that some starting materials are not sterile as they have inherent microflora that will not be eradicated by processing or manufacturing steps. Formulate the microbiological specifications for these finished products to specify the maximum allowable level. Specify the absence of ‘specified microorganisms’, which would render the product unsuitable for use.
Normal microflora is to be expected in skin; in these instances, inform the treating physician and alert them to the presence of any significant microbes. Identification allows the treating physician to make informed decisions regarding suitable antibiotic coverage for the recipient. Do not name these significant microbes on the list of specified microorganisms preventing use of the tissue.
Detail the incubation and identification timeframes and the reporting procedure in the appropriate dossier section.
Section 42 – Freeze-dried skin allografts
42 Freeze dried skin allografts
The residual moisture content of human skin products that are freeze dried must be less than 5%.
Processing of grafts by freeze-drying devitalises the grafts while maintaining their structure.
Establish a maximum limit for residual water content, with the maximum level of residual moisture set at ≤ 5 %. Measure residual moisture before release of the product.
Skin grafts can be stored at ambient temperature following freeze-drying. Validate the stability of packaging for the proposed shelf life.
Section 43 – Storage and transport
43 Storage and transport
Human skin
- Immediately following collection and prior to processing, human skin must be:
- stored as follows:
- at less than 10°C in a suitable storage medium; or
- as otherwise validated by the manufacturer to prevent microbial proliferation and to ensure the quality, safety and efficacy of the human skin product manufactured using the human skin; and
- transported in a manner that ensures the relevant storage conditions specified in paragraph (a) are maintained during transportation.
Human skin products- Human skin products must be stored as follows:
- at less than minus 40°C for not more than 5 years after collection of the human skin; or
- at 2°C to 8°C for not more than 14 days after collection of the human skin; or
- if stored in greater than 75% glycerol solution—at 2°C to 8°C for not more than 2 years after the collection of the human skin; or
- in accordance with conditions that are validated on the basis of validated data or documented evidence from relevant scientific literature, including justified time and temperature specifications, to ensure the quality, safety and efficacy of the products.
- Human skin products must be transported in a manner that ensures the relevant storage conditions specified in subsection (2) are maintained during transportation.
Transport and store (before processing commences) collected skin tissue in such a way as to maintain the quality of the allograft material, especially if the material will undergo minimal processing (viable skin allografts).
Immediately following procurement, store the recovered tissue in sterile, pre-labelled containers, filled with appropriate transport medium. Seal the containers securely, refrigerate to 2-8°C, and transport to the processing facility as soon as possible. Transportation at low temperatures prevents proliferation of most bacteria and fungi, and also maintains skin viability (if viable grafts are requested). Antibiotics can be added to the transport medium.
The specific post-processing skin storage and transport conditions detailed in this section are default storage times. Validate alternative storage conditions if specified by the manufacturer. Demonstrate that the packaging and labelling are capable of maintaining integrity and critical quality attributes of the final product, for the proposed shelf life.
Please note that in addition to TGO 109 requirements, all relevant requirements of TGO 107 (labelling of biologicals) apply to skin tissue. If further clarification is required, please contact TGA’s Biological Science Section (BSS).
Part 7 – Standard for human amnion products
Section 44 – What this Part is about
44 What this Part is about
This Part specifies requirements that must be met in the manufacture of human amnion products that have been subjected to only minimal manipulation, including requirements relating to human amnion that is collected for use in the manufacture of those products.
Unless otherwise specified, a biological to which this Part applies must meet the general requirements in Part 2 in addition to the specific requirements in this Part.
Part 7 applies to biologicals that are human amnion. This part ensures that amnion is manufactured to a minimum level and improves consistency of these therapeutic goods in Australia.
Section 45 – Application of this Part
45 Application of this Part
This Part applies in relation to human amnion products that have been subjected to only minimal manipulation.
Minimal manipulation is defined by TGA here:
Section 46 – Collection
46 Collection
- Human amnion used in the manufacture of human amnion products must be collected as soon as possible after caesarean section or vaginal delivery.
- Processing of the human amnion must commence:
- within 24 hours of collection; or
- within a longer timeframe that is validated by the manufacturer to prevent microbial proliferation and to ensure the quality, safety and efficacy of the human amnion product manufactured using the human amnion.
The Council of Europe’s 4th Edition of the Guide to the quality and safety of tissues and cells for human application (2019) states that vaginal delivery of amnion rather than collection via caesarean section in a surgical environment can introduce vaginal microflora to the starting tissue. For this reason, it is suggested that this tissue undergoes sterilisation following processing. As defined earlier in TGO 109, this will require validation of the irradiation process to meet a SAL of 10-6.
Immediate collection may be as soon as practicable after caesarean section or vaginal delivery. Donor placenta/foetal membranes should be procured by medical staff at obstetrics units after caesarean section. Amnion could be contaminated by normal vaginal flora during vaginal delivery; therefore, procurement under aseptic conditions after elective caesarean section is to be preferred. Staff undertaking procurement must be dressed appropriately for the procedure to minimise the risk of contamination of the procured tissue and any hazard to themselves.
Procurement under aseptic conditions after elective caesarean section is preferred, as amnion collected following vaginal delivery can be contaminated by normal vaginal flora.
Sterilise the processed product, if procured during vaginal delivery. Use a validated sterilisation method to achieve a SAL of 10-6 and ensure that any contaminating microflora is eliminated.
Commence processing as soon as possible after collection, with a maximum time of 24 hours. From collection to processing, store in a suitable transport/decontamination medium to preserve the structural and biological properties of the amniotic membrane.
Section 47 – Terminal sterilisation
47 Terminal sterilisation
Human amnion products manufactured using human amnion collected following vaginal delivery must be terminally sterilised in accordance with section 14.
If the product is to be terminally sterilised, perform validation and routine control of sterilisation processes in accordance with the relevant parts of international sterilisation standards, for example:
- ISO 11135 (ethylene oxide)
- ISO 14937 (general methods)
- ISO 17665 (moist heat)
- ISO 11137 (radiation)
- ISO 14160 (chemical sterilisation)
- ISO 20857 (dry heat).
Some of these standards are relevant to the sterilisation of containers and ancillary materials used during tissue processing.
Use sterile containers. Any of the ISO standards listed above provide guidance for validating the relevant sterilisation process for containers. Containers can also be pre-purchased as sterile.
Section 48 – Dehydrated or freeze-dried products
48 Dehydrated products
The residual moisture content of human amnion products that are dehydrated or freeze-dried must be less than 15%.
Dried amnion must meet minimum levels of residual moisture to ensure stability of the product during proposed shelf life.
Residual moisture content of dehydrated or freeze-dried human amnion product must be less than 15% to ensure stability, and to prevent degradation of the product and the proliferation of any residual microorganisms.
Dehydrate amnion membranes at ambient or elevated temperatures (up to 40°C), which generally induce little change in tissue properties. Place the processed membrane on support structures before exposing to air until a final water content of ≤15% is achieved. Store the product at room temperature following packaging.
Rapidly freeze and then vacuum-dry processed membrane using a freeze-drying device. Freeze-drying amnion induces minimal changes in tissue properties. Water from the tissue is extracted through sublimation until a final water content of ≤15% is achieved. Store the product at room temperature following packaging.
Determine residual moisture for each batch of amnion using a sample that has undergone the same dehydration or freeze-drying protocol (a release test). Alternatively, validate the drying process to demonstrate process consistency.
Section 49 – Storage and transportation
49 Storage and transportation
Human amnion
- Immediately following collection and prior to processing, a human amnion must be:
- stored at less than 10°C in a suitable storage medium, or as otherwise validated by the manufacturer to prevent microbial proliferation and to ensure the quality and safety of the human amnion; and
- transported in a manner that ensures the relevant storage conditions specified in paragraph (a) are maintained during transportation.
Human amnion products
- Human amnion products must be stored as follows:
- if the products are cryopreserved—at less than minus 80°C for not more than 2 years after collection of the human amnion; or
- if the products are dehydrated or freeze-dried—at room temperature for not more than 5 years after collection of the human amnion; or
- in accordance with conditions that are validated on the basis of validated data or documented evidence from relevant scientific literature, including justified time and temperature specifications, to ensure the quality, safety and efficacy of the products.
- Human amnion products must be transported in a manner that ensures the relevant storage conditions specified in subsection (2) are maintained during transportation.
Keep the storage and transport time as short as possible between procuring placenta/foetal membranes and processing. The recommended maximum time is 24 hours. A temperature of 2-8°C should be maintained.
Maintain the temperature during transport to the manufacturing site. Temperature stability should be guaranteed:
- by the container
- by conditions of transport used
- for the time interval before processing
Place procured placenta/foetal membranes in a sterile receptacle containing a suitable transport medium or decontamination solution. Validate the composition of the transport medium and determine if antibiotics are required.
Demonstrate that the packaging and labelling can maintain integrity and critical quality attributes of the final product for the proposed shelf life.
Validate that the product meets the release specifications at the end of the proposed shelf life if the product release specifications include additional criteria linked to the product’s intended use (for example, the presence of growth factors or cytokines). Part 7 allows for alternatives to the specified shelf life if the manufacturer can provide specific validation data for their product.
‘Processing’ may include steps such as treatment with decontamination or antimicrobial solutions during transportation.
Please note that in addition to TGO 109 requirements, all relevant requirements of TGO 107 (labelling of biologicals) apply to amnion. If further clarification is required, please contact TGA’s Biological Science Section (BSS).
Annex 1: Location of requirements in dossier
| Subsection | Summary of TGO 109 requirement | Relevant dossier section/s[1]
| Summary of how requirement is met[2] | Reference documents |
|---|---|---|---|---|
| 9 | Diseases and conditions that may compromise biological | 4.1.1 Donor selection (Medical and Social history) 4.1.2 Donor blood sampling and testing 4.1.3 Donor evaluation and management | ||
| 10(1)-10(2) | Critical materials used in collection of HCT materials and manufacturing process | 4.2.3 Control of materials and equipment | ||
| 11 | Microbial contamination minimisation strategy | 4.2.2. Description of manufacturing process and process controls 4.2.4. Critical steps and intermediates 4.2.5. Validation of the manufacturing process | ||
| 12 | Samples for bioburden testing | 4.1.4. Collection 4.2.4. Critical steps and intermediates 4.2.5. Validation of the manufacturing process | ||
| 13 | Bioburden testing requirements | 4.4.1. Release specifications 4.4.5. Justification of specifications | ||
| 14 | Final sterilisation validation | 4.4.1. Release specifications 4.4.5. Justification of specifications 4.2.4. Critical steps and intermediates 4.2.5. Validation of the manufacturing process | ||
| 15 | Collection from deceased donors - timeframes | 4.1.4. Collection of starting material | ||
| 16(1)-16(3) | Storage and transportation – collected HCT materials | 4.1.4. Collection of starting material 4.7. Transportation | ||
| 16(4)-16(5) | Storage and transportation – biological (final product) | 4.5. Stability and storage 4.7. Transportation | ||
| 17 | Containers of biological (final product) | 4.4.6. Containers | ||
| 20 | Musculoskeletal tissue – collection timeframes for deceased donors | 4.1.4. Collection of starting material
| ||
| 21(1)-21(2) | Musculoskeletal tissue – bioburden sampling and testing for tissue not subjected to processing | 4.1.4. Collection 4.4.1. Release specifications 4.2.4. Critical steps and intermediates | ||
| 21(3)-21(4) | Musculoskeletal tissue – bioburden sampling and testing for tissue not subjected to further processing | 4.1.4. Collection 4.2.2. Description of Manufacturing process 4.4.1. Release specifications 4.2.4. Critical steps and intermediates 4.2.5. Validation of manufacturing process | ||
| 22 | Musculoskeletal tissue – residual calcium specification for demineralised bone | 4.2.5. Validation of manufacturing process 4.4.1. Release specifications | ||
| 23 | Musculoskeletal tissue – residual moisture specification for freeze-dried products | 4.2.5. Validation of manufacturing process 4.4.1. Release specifications | ||
| 24 | Musculoskeletal tissue – final product shelf life, storage and transportation | 4.5. Storage and Stability 4.7. Transportation | ||
| 27 | Cardiovascular tissue – tissue not subjected to a bioburden reduction process | 4.1.4. Collection 4.2.2. Description of Manufacturing process 4.2.4. Critical steps and intermediates | ||
| 28 | Cardiovascular tissue – tissue subjected to a bioburden reduction process | 4.1.4. Collection 4.2.2. Description of Manufacturing process 4.2.4. Critical steps and intermediates | ||
| 29 | Cardiovascular tissue – assessment of heart valve for competency | 4.2.2. Description of Manufacturing process 4.2.4. Critical steps and intermediates | ||
| 30 | Musculoskeletal tissue – final product shelf life, storage and transportation | 4.5. Storage and Stability 4.7. Transportation | ||
| 33 | Ocular tissue – collection timeframes | 4.1.4. Collection of starting material | ||
| 34 | Ocular tissue – storage timeframes and conditions | 4.2.2. Description of Manufacturing process 4.2.4. Critical steps and intermediates 4.5. Stability and Storage | ||
| 35 | Ocular tissue – normothermic cornea microbial contamination testing requirements | 4.2.3. Control of critical materials 4.4.1. Release specifications 4.6. Labelling and release documentation 4.7. Transportation | ||
| 36 | Ocular tissue - containers | 4.4.6. Containers | ||
| 37 | Ocular tissue – examination and evaluation | 4.4.1. Release specifications 4.4.5. Justification of specifications | ||
| 40(1) | Skin tissue – bioburden sampling prior to packaging | 4.1.4. Collection
| ||
| 40(2) | Skin tissue – commencement of processing within 24 hours of collection | 4.1.4. Collection 4.2.2. Description of Manufacturing process 4.2.4. Critical steps and intermediates | ||
| 41(1) | Skin tissue – bioburden sampling and testing | 4.1.4. Collection 4.2.2. Description of Manufacturing process 4.4.1. Release specifications 4.2.4. Critical steps and intermediates 4.2.5. Validation of manufacturing process | ||
| 42 | Skin tissue – residual moisture specification for freeze-dried products | 4.2.5. Validation of manufacturing process 4.4.1. Release specifications | ||
| 43(1) | Skin tissue – storage and transportation before processing | 4.1.4. Collection 4.2.4. Critical steps and intermediates | ||
| 43(2)-43(3) | Skin tissue – storage, shelf-life and transportation after processing | 4.5. Storage and Stability 4.7. Transportation | ||
| 46 | Amnion – collection and pre-processing timeframes | 4.1.4. Collection 4.2.4. Critical steps and intermediates | ||
| 47 | Amnion – terminal sterilisation requirements | 4.1.4. Collection 4.2.2. Description of Manufacturing process 4.2.5. Validation of manufacturing process 4.4.1. Release specifications | ||
| 48 | Amnion- residual moisture specification for dried tissue | 4.2.5. Validation of manufacturing process 4.4.1. Release specifications | ||
| 49(1) | Skin tissue – storage and transportation before processing | 4.1.4. Collection 4.2.4. Critical steps and intermediates | ||
| 49(2)-49(3) | Skin tissue – storage, shelf-life and transportation after processing | 4.5. Storage and Stability 4.7. Transportation |
Footnotes
- Suggested dossier location; actual location of information may vary depending on the nature of the product, but must be defined under this heading.
- Only a very brief summary is required, the entire dossier will be evaluated.
Page history
Title changed from 'ARGB Appendix 13 - Guidance on TGO 109: Standards for Biologicals-General and Specific Requirements' to 'Understanding rules for manufacturing biologicals ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Addition of Annex 1.
Original publication.
Title changed from 'ARGB Appendix 13 - Guidance on TGO 109: Standards for Biologicals-General and Specific Requirements' to 'Understanding rules for manufacturing biologicals ' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Addition of Annex 1.
Original publication.