Understanding good manufacturing practice (GMP) for registered medicinal gases
This guidance is for manufacturers of registered medicinal gases to understand our interpretation of the Pharmaceutical Inspection Co-operation Scheme (PIC/S) guide to GMP for medicinal products.
Purpose
This guidance is for manufacturers of registered medicinal gases. It explains our interpretation and expectations for compliance with specific sections of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products PE009-13 (PIC/S Guide to GMP).
TGA has adopted the PIC/S Guide to GMP, PE009-13 (excluding Annexes 4, 5 and 14), taking effect on 1 January 2018 with a 12-month transition plan.
Scientific guidelines
TGA regulates medicinal gases
Medicinal gases are:
- classified as medicines under the Therapeutic Goods Act 1989 (the Act)
- required to comply with:
- Therapeutic Goods (Manufacturing Principles) Determination
- PIC/S Guide to Good Manufacturing Practice for Medicinal Products PE009-13 (PIC/S Guide to GMP).
Exemptions from regulation
Bulk liquefied medical gases
Bulk liquefied medical gases are exempt from the operation of Part 3-3 of the Act, under Schedule 7, Item 17 (Manufacturing of therapeutic goods).
Medical oxygen cylinders
Oxygen cylinders that have been decant filled, transfilled or cascade filled by a hospital; or an ambulance, fire or rescue service are exempt from the operation of Part 3-3 of the Act, under Schedule 7, Item 20.
Related information
TGA guidance relevant to medicinal gas manufacturing includes:
- Manufacturing medicines
- Data Management and Data Integrity (DMDI) Guidance
- Guidance on release for supply
- Uniform recall procedure for therapeutic goods
Other useful guidance:
Sections of PE009-13 that apply
All relevant clauses of the PIC/S Guide to GMP apply to the manufacture of medicinal gases; however, specific guidance regarding the interpretation of certain GMP requirements is provided herein. Where this guide does not provide any commentary regarding interpretation, it is expected that the manufacturer fully comply with those requirement(s), without further interpretational guidance.
In general, you should follow the principles of Part I of PE009-13, and in addition, all annexes relevant to the manufacture of medicinal gases.
You should meet the requirements of the following Annexes:
- Annex 6 (Manufacture of Medicinal Gases)
- Annex 11 (Computerised Systems)
- Annex 15 (Qualification and Validation)
- Annex 20 (Quality Risk Management) NB this is a voluntary annex; however, the principles outlined in the annex are applicable to the manufacture of therapeutic medicinal gases.
Quality management: chapter 1
Quality management applies to the manufacture of all therapeutic goods to which the PIC/S guide to GMP (PE009-13) applies, including medicinal gases.
Terminology for quality management
The following quality management terms apply to manufacturing of medicinal gases:
Pharmaceutical Quality System
The latest PIC/S Guide to GMP, replaces the terminology 'Quality Management System' with 'Pharmaceutical Quality System' (PQS). This is in line with:
- ICH Q10 global harmonisation efforts
- PIC/S harmonisation efforts
- aligning the GMP guide with contemporary principles of quality systems management
The new terminology better reflects the specific design elements and requirements for a quality system used to manage the manufacture of medicinal products.
The PQS approach described is applicable to the manufacture of all therapeutic goods to which the PE009-13 applies.
As medicinal gas manufacture is only a small component of most Australian gas manufacturing operations it is not necessary for manufacturer's documents to specifically refer to a Pharmaceutical Quality System.
Manufacturing authorisation
The term 'manufacturing authorisation', generally refers to the licence to manufacture therapeutic goods issued by the TGA to domestic manufacturers. For manufacturers located overseas, this would refer to the certificate of GMP compliance issued following an inspection.
Marketing authorisation
Marketing authorisation is the approval given to supply a therapeutic good in Australia, and, in most cases, involves entry on the Australian Register of Therapeutic Goods (ARTG).
The marketing authorisation includes the details of the product in the Australian Register of Therapeutic Goods (ARTG), as well as all other matters in relation to product registration, listing or inclusion agreed in writing between the TGA and the sponsor, and any other requirements imposed by a relevant Delegate of the Secretary upon ARTG entry.
Examples of regulatory requirements include, but are not limited to:
- compliance with standards and registered formulations
- special storage and transportation conditions
- shelf life
- packaging and labelling
- batch release testing requirements
- manufacturers are responsible for ensuring their PQSs are designed and operated to ensure all relevant requirements of the marketing authorisation are observed during the manufacture of therapeutic goods
- holder of the marketing authorisation
The holder of the marketing authorisation is the product sponsor.
Change management
Regulated changes
Regulated changes include any manufacturing change that affects the registered product details.
Regulated changes:
- are included as requirements for the marketing authorisation of therapeutic goods
- require an application to vary the marketing authorisation
Please refer to Australian Regulatory Guidelines for Prescription Medicines (ARGPM).
These requirements are mandatory and are in addition to the requirements of the PIC/S Guide to GMP (PE009-13).
Change control applies to all GMP-related activities
Change control applies to all GMP-related activities undertaken by a manufacturer, not just to validation activities (clause 1.4 xii, xiii).
Change control and risk assessment requirements within the PIC/S Guide to GMP (PE009-13) apply to both:
- regulated changes
- other changes that have the potential to impact product quality
Verify all implemented changes for effectiveness through a change control process, including, but not limited to, changes to existing:
- processes
- systems
- facilities
- equipment
- products
- documents
The effort and extent of change control processes should be:
- commensurate with the nature of the change
- based on risk management principles
Verify that all changes implemented are effective following implementation.
Managing deviations
Manage deviations and other similar events according to the PIC/S Guide to GMP (clause 1.4 xiv). The guide clarifies the expectations for investigating deviations, including:
- adequate root-cause-analysis
- identification of corrective and preventative actions
Monitor and assess the effectiveness of such in line with quality risk management (QRM) principles.
Release for supply
Release for further processing (RFFP) and release for supply (RFS) are conducted by authorised persons. For more guidance see:
The manufacture of medicinal gases is the subject of Annex 6 of the PIC/S guide to GMP.
Unique features of medicinal gas manufacture
The manufacture of medicinal gas products differs from that of other medicinal products. These differences necessitate altered expectations for RFFP and RFS.
Features of medicinal gas manufacture:
- use of re-useable packaging (gas cylinders)
- bulk product manufacture in non-GMP licensed facilities
- packaging testing in non-GMP licensed facilities
- absence of stability programs
- dispersed location of multiple manufacturing sites
There are often multiple manufacturing sites across the country for one medical gas product. It is not practical to have a centralised manufacturing site because logistical constraints make it desirable for the bulk production sites and cylinder-filling sites to be located as close as possible to the end users.
The number and dispersed location of manufacturing sites make it necessary for authorised persons to be allowed to conduct RFFP and RFS from locations that are remote from the actual manufacturing site.
Process flowchart for medicinal gas RFS
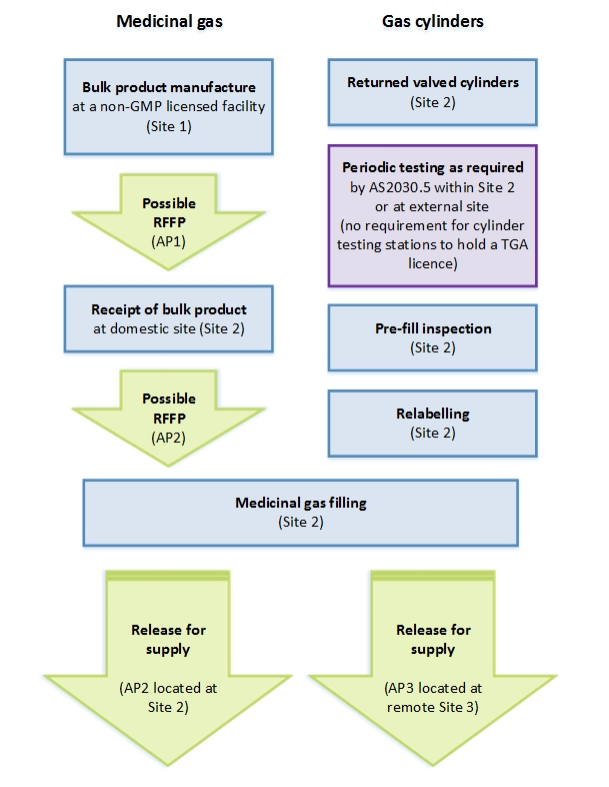
{kind=link}
Flow chart showing the medicinal gas manufacturing and cylinder filling process across multiple sites. The process is divided into two parallel streams: 'Medicinal gas' on the left and 'Gas cylinders' on the right.
Left stream starts with 'Bulk product manufacture at a non-GMP licensed facility (Site 1)', followed by a possible RFFP (AP1), then 'Receipt of bulk product at domestic site (Site 2)', another possible RFFP (AP2), leading to 'Medicinal gas filling (Site 2)'. This branches into two release paths: 'Release for supply (AP2 located at Site 2)' and 'Release for supply (AP3 located at remote Site 3)'.
Right stream shows cylinder preparation steps at Site 2: 'Returned valved cylinders', 'Periodic testing as required by AS2030.5 within Site 2 or at external site (no requirement for cylinder testing stations to hold a TGA licence)', 'Pre-fill inspection', and 'Relabelling'. These steps feed into the same 'Medicinal gas filling (Site 2)' process as the left stream.
All process boxes are shown in blue except for the RFFP and Release steps which are in green, and the periodic testing note which is in purple.
Bulk manufacture
The site of bulk manufacture, Site 1, will typically be one of the following:
- a non-GMP licensed Australian facility
- an overseas facility with or without TGA GMP clearance
GMP licence not required
Bulk manufacture of medicinal gases does not need to be in a GMP-licensed facility (item 17, Schedule 7 of the Therapeutic Goods Regulations 1990).
Analysis at Site 1
Site 1 performs analysis for identity and purity and the subsequent generation of a certificate of analysis of the bulk product.
Quality agreement between sites 1 and 2
When sites 1 and 2 operate under separate quality management systems, there should be a quality agreement between the two facilities to ensure that the manufacturer of the bulk product notifies the manufacturer at Site 2 of any:
- significant deviations from the agreed production process for the bulk gas
- out-of-specification results
- non-compliance with quality management systems
- investigations
- complaints
- other matters that may potentially affect product quality adversely
Transportation and RFFP
Transportation of bulk product to Site 2 can be before or after RFFP of the bulk product.
RFFP of bulk product
You should ensure that the quality of the delivered bulk gas is acceptable (clause 21 of Annex 6 of the PIC/S guide to GMP).
RFFP of the bulk product can be done:
- by an authorised person (AP1) at the bulk manufacturing site, despite this site not being GMP-licensed
- by an authorised person (AP2) at the GMP-licensed Site 2
This RFFP is limited to:
- checking for the availability of a certificate of analysis
- verifying:
- the product's identity
- the purity is within pre-defined specifications
We do not require the authorised person to do more than this because the bulk manufacturing facility is usually not GMP-licensed or GMP-cleared.
Quality of cylinders
Empty valved medicinal gas cylinders are:
- returned to an Australian GMP-licenced medicinal gas filling facility
- refilled and re-used repeatedly
Periodic testing of returned cylinders
Requirements for periodically inspecting and testing gas cylinders are specified in AS 2030.5 (see SAI Global) Gas cylinders – filling, inspection and testing of refillable cylinders.
These inspections and testing:
- do not need to be in a GMP-licensed facility
- can be at Site 2 or external to Site 2
- are performed at a certified gas cylinder test station, which are required to
- satisfy the requirements of PIC/S guide to GMP
- be audited annually for re-certification purposes be an external qualified certifier
- shall be in accordance with AS2337.1 and AS2337.3 (see SAI Global):
- gas cylinder test stations - General requirements, inspection and tests - Gas cylinders
- gas cylinder test stations - Transportable gas cylinders - Periodic inspection and testing of composite gas cylinders (ISO 11623:2002, MOD)
Third party gas cylinder test stations
If you engage third party gas cylinder test stations:
- use a contractor qualification process
put in place a quality agreement that clearly defines GMP responsibilities and obligations of the contractor (clause 28 of Annex 6 of the PIC/S guide to GMP).
Microbial monitoring program
Water used in the hydrostatic pressure testing of gas cylinders must be at least of drinking water quality (clause 26 of Annex 6 of the PIC/S guide to GMP).
If you use an external contractor to monitor water quality, you need to have a quality agreement with the contractor to ensure immediate reporting to Site 2 of anomalous or out-of-specification results that might have the potential to impact product quality adversely.
Pre-fill inspection of cylinders
Prior to filling, inspect all returned cylinders for assessment of suitability for re-use in accordance with quality and functionality criteria specified in clause 30 of Annex 6 of the PIC/S guide to GMP.
Labelling
Label each container as described in clause 37 of Annex 6 of the PIC/S guide to GMP.
Filling and post-filling of cylinders
Use gas cylinders that have passed the quality inspection and fill with medicinal gas from the bulk product in accordance with the marketing authorisation.
There should be checks that each container is properly filled (clause 34 of Annex 6 of the PIC/S guide to GMP).
You may define a batch of liquefied medicinal gas product, by specifying a period of continuous filling.
Release for supply of medicinal gases
The RFS authorised person should have a thorough knowledge of the production and control of medicinal gases. So long as the person meets the criteria for being authorised to perform release for supply of medicinal gases, the authorised person can be a trained independent operator.
The authorised person must review appropriate documentation and release the product:
- at the medicinal gas filling facility, Site 2 (authorised person 2, AP2)
- at Site 3 (remote authorised person, AP3)
Using a remote authorised person for release for supply
If you use a remote authorised person (AP3):
- Site 2 and Site 3 should operate under one quality management system
OR
- there should be a GMP agreement between the two facilities defining responsibilities in regard to release for supply
Documentation for RFS
For RFS of medicinal gases, include documentation to demonstrate:
- appropriate pre-fill inspections of cylinders have been conducted and all cylinders used in the batch are acceptable for use
- the medicinal gas has been tested according to its specification
- each cylinder in the batch is appropriately labelled with its batch number
- appropriate checks have been conducted to ensure all containers have been filled
- each cylinder within the batch has been tested for leaks and fitted with tamper evident seal
Verifying the quality of starting materials
For release for supply, the authorised person is required when possible to verify the original quality characteristics of starting materials. For medicinal gas products, it is not possible to do this for all starting materials because the quantity of bulk product, packaging and its accessories are not:
- allocated to a specific finished product batch
- reconciled for a batch (e.g. product labels, valves, dust caps etc.)
This is due to:
- the characteristics of the bulk product (cryogenic liquid)
- losses due to natural vaporisation
- the re-use of packaging and most items associated with it
Ensuring traceability
You are expected to be able to trace:
- gas cylinders
- contents
You are not expected to be able to trace other components of medicinal gas products. This is because all accessories stay on the gas cylinders unless unfit for use (e.g. product labels, valves, dust caps, etc.), at which point they are replaced at the appropriate manufacturing step. The accessories are not allocated to a specific batch.
No ongoing stability program
Unless specified otherwise in the marketing authorisation, medicinal gases do not have a nominated shelf life, so you do not need an ongoing stability program.
Senior management responsibilities for GMP and quality management
Particular emphasis is placed on the roles and responsibilities of senior management who have ultimate control over manufacturing facilities and activities (new clauses, including clause 1.5).
Senior management hold the responsibility:
- to ensure that the manufacturing activity is managed appropriately
- to ensure adequate resources are available (human, financial and physical)
- for the effectiveness of the PQS
- for implementing an effective PQS
- undertaking an active role in the support, development and implementation of the PQS
Management reviews
Management reviews (clause 1.6) are:
- a basic quality system element
- designed to inform the management group of the effectiveness of the PQS
- important in resolving issues and managing risks by:
- escalating concerns
- enabling senior management support
TGA's basic expectations, based on ICH Q10 principles, are that the management review system should include:
- results and findings of:
- regulatory inspections
- audits and other assessments
- commitments made to regulatory authorities
- periodic reviews of management quality, which can include:
- measures of customer satisfaction, such as product quality complaints and recalls
- conclusions of process performance and product quality monitoring
- the effectiveness of process and product changes, including those arising from corrective action and preventive actions
- follow-up actions from previous management reviews
The management review system should also identify appropriate actions, such as:
- improvements to manufacturing processes and products
- provision, training and/or realignment of resources
- capture and dissemination of knowledge
Management review of the Pharmaceutical Quality System
Management are to formally review the PQS on a periodic basis by:
- measuring the achievement of PQS objectives
- assessing the performance indicators used to monitor the effectiveness of processes within the PQS, such as:
- complaint, deviation, corrective and preventative actions (CAPA) and change management processes
- self-assessment processes, including risk assessments, trending, and audits
- external assessments, such as regulatory inspections and findings, and customer audits
Monitoring by managers can include both internal and external factors impacting the PQS, such as:
- emerging regulations, guidance and quality issues
- innovations that might enhance the PQS
- changes in business environment and objectives
- changes in product ownership
Frequency of management reviews
TGA inspectors would generally expect reviews to be conducted at least annually (clause 1.6). However, management reviews may be performed more frequently for:
- new operations
- sites that have not previously performed management reviews
- sites aligned with corporate policies
- sites where the initial management review identifies a number of issues that require rectification
- sites with larger and more diverse manufacturing operations
Quality Manual development
Write and maintain a Quality Manual, or equivalent document (clause 1.7).
Describe the PQS in your quality manual, or equivalent, including:
- the quality policy
- identification of the PQS processes
- PQS process sequences, linkages and interdependencies. Process maps and flow charts can be useful tools to facilitate depicting PQS processes in a visual manner
- management responsibilities within the PQS
Product distribution expectations
When you distribute products:
- minimise any risk to product quality
- take account of 'good distribution practice' (GDP) (clause 1.8 (ix))
TGA does not currently inspect the wholesale distribution of therapeutic goods that have been released for supply.
Sponsors and manufacturers have shared responsibility to ensure the quality of medicinal gases are maintained throughout their shelf life, by:
- ensuring products are stored, distributed and subsequently handled appropriately
- establishing quality or technical agreements to clearly identify responsibilities, where manufacturer(s) and sponsor are separate entities
Transportation between manufacturing sites
TGA inspections evaluate the transport conditions between sites of manufacture for:
- Bulk gas
- packed medicines moved between sites of manufacture
In these circumstances, clause 1.8 (ix) would apply.
Product Quality Reviews (PQRs)
PQRs must be performed for all compressed medicinal gases that are filled on-site.
PQRs are not required for bulk liquefied gases.
There are no requirements to review the stability monitoring programme where expiry dates are not required on the individual batch records or cylinder labels.
Medical air production:
- is generally:
- only subject to Part I of the PIC/S Guide to GMP due to the nature of its manufacture
- is not subject to Part II of the PIC/S Guide to GMP
- needs to be included in the PQR, where the manufacture starts with the compression of air at the filling site
Supply chain traceability for active substance gases
Manufacturers of medicinal gases should have a clear understanding of:
- the approved suppliers of active substance gases, for each entity
- their responsibility in the supply chain between the site of manufacture of the bulk gas and receipt (clause 1.10(i))
Supply chains should:
- be adequately secure
- ensure bulk gases are transported under appropriate conditions
- be mapped
- have all identified risks managed following the principles of quality risk management
Frequency of PQRs
Perform a full PQR on at least a yearly basis, reviewing all relevant elements of clause 1.10.
It may be acceptable to perform a full PQR on a two yearly basis, when:
- very few batches of one product are manufactured in one year
- no manufacturing takes place
- a scientifically justified rationale is documented
Manufacturers and sponsors are expected to maintain vigilance over elements of clause 1.10 that do not directly relate to manufacturing activities.
For example, during periods where very few, or no, batches of one product are manufactured, recalls and complaints may provide information about products in the market that needs to be recorded in the PQR.
Grouping of products for PQR
Grouping of products is when one PQR is prepared for a group of products. Grouping for the preparation of PQRs is acceptable for medicinal gases.
Batches to be included in a PQR
PQRs are to include all batches for which manufacture:
- commenced during the current PQR reporting period
- was terminated, delayed or failed
When grouping is applied, all batches of all products in each group are expected to be included in the PQR.
Shared responsibility for PQRs between manufacturers and the sponsor
Manufacturers and sponsors share responsibility for:
- preparing the PQR
- designing and implementing effective systems to ensure that PQR reports and relevant data are supplied, compiled and reviewed
- establishing technical agreements to clearly define each party's responsibilities in relation to PQRs
Sponsors are also expected to have access to the PQRs, to ensure product compliance with the marketing authorisation.
Quality Risk Management
Quality risk management is mandatory
You must have an operational quality risk management system to ensure that:
- the evaluation of a risk to product quality is based on a sound, scientific basis and
- risk assessments are appropriately documented (clauses 1.12 and 1.13)
Annex 20 is voluntary and provides guidance only on quality risk management tools that may be applied by a manufacturer when assessing the risk to product quality.
Personnel: chapter 2
Senior management responsibilities for personnel
Particular emphasis is placed on the roles and responsibilities of senior management who have ultimate control over manufacturing facilities and activities in new clauses of PE009-13 (including clause 2.1 & 2.4). Senior management are accountable for ensuring appropriate resources are available to support the relevant manufacturing activities.
Personnel qualifications
Necessary qualifications for staff and consultants
'Necessary qualifications' means having the education, training, experience and skills, or any combination of these elements, that will ensure that staff can perform assigned duties and functions at an acceptable level (clause 2.1).
Similar records should be available for consultants used for the manufacture of medicinal gases stating the name, address, qualifications, and type of service provided by these consultants (clause 2.23).
Expectations for training and language
All personnel are to be aware of the principles of Good Manufacturing Practice that affect them and receive initial and continuing training, including hygiene instructions, relevant to their needs.
Relevant training information can be found in the PIC/S guide to GMP Part 1 clause 2.10 to 2.14.
Training requirements
Define and document procedures for all training requirements, including initial and ongoing training.
Generate and keep training records for all staff trained.
Trainers
Ensure any person conducting training and assessment is appropriately:
- trained
- qualified
- experienced in the subject matter
Trainees
Provide and record appropriate training and assessment to:
- all people affected by significant change in the PQS, for example, when SOPs or equipment changes
- people who have a direct bearing on quality outcomes, including:
- senior management
- contractors
- casual employees
Language requirements
Manufacturers are to:
- define language requirements or standards
- ensure personnel are proficient in the required language for their allocated tasks, particularly for documenting and recording tasks
- document procedures to overcome identifiable language deficiencies
Personnel hygiene
Manufacturers are responsible for ensuring that the health and hygiene practices of the operators involved in manufacture do not affect the quality of the gases manufactured.
Every person entering the manufacturing areas is expected to (clauses 2.18 & 2.19):
- wear personal protective equipment appropriate to the operations to be carried out
- be prohibited, in production and storage areas, from:
- eating, drinking, chewing or smoking
- storing food, drink, smoking materials or personal medication
- drinking water in closed containers is permissible
Due to the nature of manufacturing operations, and the minimal risk to product quality, a number of clauses in Chapter 2 of the PIC/S Guide to GMP pertaining to personnel hygiene are not applicable to the manufacture of medicinal gases:
- Personal hygiene, clauses: 2.15, 2.17, 2.20 & 2.22
Premises and equipment: chapter 3
Due to the method of manufacture of medicinal gas, not all of the requirements of Chapter 3 of the PIC/S Guide to GMP are applicable.
Outlined below are the sections of Chapter 3 of the PIC/S guide to GMP that are either:
- not applicable
- superseded by information in Annex 6
| Clause | Interpretation |
|---|---|
| 3.2 | Replaced by specific requirements in Annex 6: clause 8 |
| 3.3 | Lighting should be adequate for the tasks being performed by the filling operators. Temperature is only relevant when calculating the settled pressure of the gas. Ventilation of the gas filling area must be adequate for safety reasons in the event of gas leakage. Humidity is not relevant to closed filling systems. |
| 3.4 | Not applicable |
| 3.6 | Not applicable |
| 3.7 | Applicable (replaced by specific requirements in Annex 6: clause 8) |
| 3.9-3.14 | Not applicable |
| 3.22 | Not applicable |
| 3.26 | Batch specific testing and full cylinder analysis against product specifications (i.e. BP/EP/USP monographs), should be performed using equipment that is appropriately calibrated, maintained and where applicable utilise validated systems. |
| 3.29 | Not applicable |
| 3.31 | Not applicable |
| 3.33 | Not applicable |
| 3.43 | Not applicable |
Environmental controls for filling operations
Manufacturers are responsible for ensuring that the environmental conditions during manufacture are suitable (clause 3.3). TGA's basic expectations for environmental controls are that:
- lighting should be adequate for the tasks being performed by the filling operators
- temperature controls are only relevant when calculating the settled pressure of the gas
- ventilation of the gas filling area must be adequate for safety reasons in the event of gas leakage
- humidity control is not relevant to closed filling systems
Pest control
The requirements relating to pest control (clause 3.4) are not relevant to medicinal gas manufacture.
Precautions against cross-contamination
The prevention of cross-contamination events should be assured by the use of robust engineering controls for filling equipment. The principle of clause 3.6 applies to the manufacture of medicinal gases in that adequate precautions should be taken to prevent cross-contamination.
Annex 6 contains information about the required controls.
Design and control of production areas
Production areas for the manufacture of medicinal gas:
- do not require clean-rooms, filtered air environments or similar controls (clauses 3.9 – 3.14)
- do not normally involve the preparation or exposure of starting materials or primary packaging materials
Cylinder storage and sampling
Medicinal gas manufacture:
- has basic requirements relating to storage capacity (clause 3.18) and design (clause 3.19)
- has no requirement for a separate sampling area for staring materials (clause 3.22)
Annex 6 contains information about cylinder storage.
Printed packaging material storage
Cylinder product labels can be:
- changed at several points in the manufacturing process
- made available at multiple areas of the facility such as:
- test shop
- filling ramps
- maintenance area
For the purposes of clause 3.25, the minimum requirements to demonstrate 'safe and secure storage' of cylinder product labels are to:
- keep bulk quantities of cylinder product labels secure in a restricted storage area to exclude unauthorised access
- provide secure separate locations for quarantined and released cylinder product labels (clause 3.21)
- keep only a minimal number of approved cylinder product labels in the production areas, e.g. enough for one week's production
- store cut labels in separate, closed containers to avoid mix-ups (clause 5.41)
- provide secure, lockable storage of released-to-production cylinder product labels at the end of shifts in all areas of the facility
Quality Control laboratories
Quality Control laboratories should generally be separated from production (clause 3.26); however, where testing activities do not pose a risk to product quality they may be conducted within the production environment.
Perform batch specific testing against the product specification using equipment that is appropriately calibrated, maintained, and where applicable, utilise validated systems.
Detailed requirements for quality control of medicinal gases are outlined in the relevant 'Quality Control' section of Annex 6 clauses 39-44.
Ancillary areas
Medicinal gas manufacturers are:
- required to provide toilets appropriately located for the number of users
- not required to provide facilities for changing clothes and hand washing (not GMP critical requirements, clause 3.31)
Cleaning and sanitisation
The PIC/S Guide to GMP (PE009-13) contains limited detail on requirements for cleaning and sanitisation specifically related to medicinal gas manufacturers.
Closed pressurised systems and equipment used for medicinal gas manufacture, only needs cleaning between batches if exposed to a contaminant.
Manufacturers should retain appropriate records when cleaning pressurised systems and equipment prior to use in commercial production, for example after:
- post qualification and commissioning
- any closed system breaches to the system's integrity
(Annex 6 clause 15, Annex 15 clause 3.1).
Process water
Distilled or deionised water is not normally used in the production of medicinal gases.
The hydrostatic testing of cylinders can use town (drinking) water (Annex 6 clause 26):
- Use a risk-based approach when considering the need to sanitise water pipes within the hydrostatic test station, particularly for processes that recirculate water through multiple testing events
- Document:
- written procedures describing microbiological testing of process water used in the hydrostatic testing of cylinders
- action limits for microbiological contamination, and measure
Documentation: chapter 4
Retention of batch documents
Keep batch documentation for products:
- with an expiry date, for whichever is the longer period, of:
one year after expiry of the batch to which it relates
OR
- at least five years after release for supply of the batch by the authorised person
- with no expiry date:
- for at least 6 years after completion of manufacture of the goods, in accordance with section 20 of the Therapeutic Goods Regulations 1990
Production records
Specific details for the content of batch records for medicinal gases can be found in the PIC/S Guide to GMP: Annex 6 clause 17 & 30.
The following clauses of Chapter 4 of the PIC/S Guide to GMP apply to each batch of medicinal gas filled:
- packaging instruction (clause 4.19)
- batch processing record (clause 4.20)
- batch packaging record (clause 4.21)
Clauses of Chapter 4 of the PIC/S Guide to GMP that do not apply to medicinal gas manufacture include:
- manufacturing formula (clause 4.17)
- processing instructions (clause 4.18)
Control of cylinder product labels
For starting materials and packaging materials (clauses 4.21-4.27), including cylinder product labels, provide written procedures and records, for:
- receipt
- storage
- sampling
- testing
- release/rejection
For cylinder product-labels, provide written procedures for how to:
- quarantine
- inspect
- release
- issue to production
- control
It is not mandatory to reconcile cylinder product label usage for each batch at the filling stations.
Establish processes to:
- control changes in product labels
- ensure all obsolete labels are removed from the site and destroyed where a ‘hard label’ change is required
Maintaining policies, procedures and records
Provide appropriate documentation to demonstrate compliance with each element of clause 4.29.
Where reference is made to environmental monitoring, this is normally limited to monitoring of specific processing or storage conditions, for example, temperature, where required by the process.
Further guidance is provided in Annex 6 clause 17 & 18.
Log-books
Use logbooks for all analytical equipment.
Where there is adequate traceability of activities, logbooks are not required, for:
- product-specific (dedicated) equipment
- areas utilised for production
Signature list
For individuals who complete GMP documentation, maintain a signature list that includes:
- names
- signatures
- initials
The signature list is the key reference when providing traceability between manual signatures used on documents and the individuals who completed them.
Production: chapter 5
The following guidance, on demonstrating compliance with the PIC/S guide to GMP has been formulated in relation to the following:
- in Australia, the production of bulk liquified gases is exempt from compliance with the PIC/S Guide to GMP
- the filling/manufacture of medical air starts with the compression of air and the medical air compressors, and all filling operations, should comply with Part 1 of the PIC/S Guide to GMP
Yields and reconciliation of quantities
We expect manufacturers of medicinal gas to:
- perform batch label reconciliation when cylinders have printed batch labels applied
- keep records of the quantity of batch labels printed, used and destroyed
We generally do not expect manufacturers of medicinal gas to perform:
- checks on yields
- reconciliation of quantities (clause 5.8)
Loss of medicinal gas occurs during manufacturing and can be variable, even under normal operating conditions.
To determine if the amount of medicinal gas, or gas mix, in a container is the amount and type indicated by the label, it is sufficient to:
- fill to a predetermined and acceptable temperature and pressure limit, or mass target
- perform finished product testing
Working with dry materials
Clause 5.11 does not apply to the manufacture of medicinal gas, as dry materials are not normally used.
Precautions against cross-contamination
Contamination control applies to the manufacture of medicinal gases.
Relevant sections of the PIC/S Guide to GMP include:
- Clauses 5.18 and 5.19: provide general principles
- Annex 6: provides specific guidance regarding the required controls.
The manufacture of medicinal gases uses closed equipment. However, the re-use of cylinders without adequate precautions could lead to contamination by a wide variety of contaminants.
Periodic critical revalidation
Where a validated state has had no significant changes, a review with evidence that facilities, equipment and processes meet the prescribed requirements fulfils the need for periodic critical revalidation under clause 5.24.
The evidentiary review could be performed in conjunction with the PQR process.
Labelling of starting materials
Starting materials for medicinal gases include:
- empty gas cylinders (reused packaging material)
- cylinder product labels (not generally replaced each filling cycle)
- bulk gases (liquified or compressed gases stored in dedicated storage or buffer tanks)
Due to the unique product format of medicinal gases, not all requirements in clause 5.29 for labelling starting materials during storage apply.
Labelling requirements for starting material during storage that do apply to medicinal gases include:
- the designated name of the product and/or the internal code reference
- where appropriate, an expiry date or a date beyond which retesting is required
- for medicinal gas manufacture, the term 'starting material' also refers to the empty gas cylinder
Labelling requirements for starting material during storage that do not apply to medicinal gases include:
- a batch number given at receipt
- where appropriate, the status of the contents (e.g. quarantine, on test, released, rejected).
Dispensing of starting materials
Dispensing activities (clauses 5.32-5.34):
- are not applicable to the manufacture of medicinal gases.
Medicinal gas manufacture does not encompass dispensing of starting materials for later use in batch production. Bulk product is filled directly into the cylinders using closed equipment.
Intermediate and bulk products
The requirements for area clearance (clause 5.35), and intermediate and bulk storage (clause 5.36) are applicable to the manufacture of mixed medicinal gases only.
Packaging materials
The expectations to demonstrate compliance with clauses 5.41-5.43 are outlined in the 'printed packaging material storage' and 'control of cylinder product labels' sections of this guidance.
Products and batch details to be displayed
For the purposes of demonstrating compliance with clause 5.46 there is no requirement to display the name and batch number of the product being handled at the packaging line when the following are met:
- medicinal gas is filled on dedicated filling equipment and the equipment is labelled accordingly
- filling equipment is appropriately identified/labelled with the gas type
Electronic readers
Controls should be in place to verify electronic code readers are operating correctly, such as cylinder bar code scanners (clause 5.52).
Control of product during packaging
The basic principles outlined in clause 5.54 for on-line control of product during packaging are insufficient to adequately control the production of medicinal gases. Detailed requirements for production of medicinal gases are outlined in the relevant 'Production' section of Annex 6 clauses 19- 38.
Reconciliation of batch labels
Reconciliation of quantities for medicinal gas manufacture is limited to printed batch labels only.
Investigate discrepancies before release of the product (clause 5.56).
Destroy unused printed batch labels, and record the destruction (clause 5.57).
Reprocessing
Due to the nature of medicinal gas manufacture, the reworking of gases is not expected.
The requirements of clauses 5.62-5.64:
- do not apply where reprocessing or rework are not performed
- do apply only in situations where reprocessing or rework of gases (specifically re-labelling) occurs
Quality control: chapter 6
Reference and retention samples
Reference and retention samples (clause 6.14) for medicinal gases are not required, as outlined in Annex 6 clause 43, and therefore compliance with this clause is not expected.
Method validation
Method validation (clause 6.15) is not normally required where official test methods outlined within the default standards, (BP, Ph. Eur. or USP) are adhered to.
Ongoing stability
Ongoing stability programmes (clauses 6.26 - 6.36) for medicinal gases are not generally required, as outlined in Annex 6 clause 44. If stability requirements are detailed in the market authorisation of a product, then ongoing stability testing may be necessary.
Outsourced activities: chapter 7
Examples of outsourced activities include, but are not limited to:
contract manufacturing and analysis
- equipment maintenance and calibration services
- cylinder maintenance or testing facilities
- suppliers and manufacturers of raw materials, packaging materials and printed artwork
- provision of training and consulting services
- validation services associated with facilities, equipment, utilities, process and product design, qualification and validation
- provision of transport and logistical services for products (prior to release for supply)
- agencies that provide temporary or contract personnel
The title of chapter 7 has changed from 'Contract manufacturer and analysis' to 'Outsourced activities' in recognition of the fact that there are a number of outsourced (contracted) activities that may have a direct effect on the quality of medicinal product manufactured by a site. The previous title of the chapter restricted the extent of GMP controls to only outsourced manufacturing and testing services and thus did not appropriately manage the risk associated with other outsourced activities.
Managing outsourced activities
TGA expect manufacturers (normally 'contract givers') to:
- manage all relationships with contract acceptors in accordance with existing principles of chapter 7 of the PIS/S Guide to GMP
- assess, define and cover by a written agreement all outsourced GMP-related activities that may impact on product quality
- maintain all agreements in accordance with the PQS.
Legality of outsourced activities
The term 'legality' in clause 7.4.1 means that contract givers are responsible for making sure that the entity undertaking the outsourced activities is appropriately authorised to undertake the activity. This may be achieved by many means including ensuring that the contract acceptor:
- holds the appropriate manufacturing authorisation (licence) to undertake the specific steps in manufacture
- is nominated as being authorised to undertake the specific activity in the specific marketing authorisation of the products
- holds any necessary licenses or permits applicable to the outsourced activities
- holds the necessary accreditation related to the activities undertaken, e.g. a contract calibration company may hold NATA certification that a cylinder test station is certificated against AS2337
Monitoring the contract acceptor
The contract giver should have a system in place to measure and monitor the quality of products (or service) provided by the contract acceptor, in accordance with risk management principles.
Where quality related issues are identified, it is expected that appropriate actions are taken to address and remediate the concerns.
Records of actions taken should be recorded within the PQS.
Responsibility for review of records and results
Clause 7.5 states that the contract giver should be responsible for reviewing and assessing the records and the results related to the outsourced activities.
It is expected that the responsibility for review of the records and results to be specified by the contract should be based on the risk and nature of the service provided. For example:
- for contract service providers (e.g. contract calibration services) it would be appropriate for the contract giver to review the available records and data to ensure that the results or work provided meet the requirements of the contract giver’s quality system and procedures
- for contract manufacture and analysis, it may be appropriate for the contract giver to rely fully on the contract acceptor where an authorised representative of the contract acceptor, (e.g. quality manager), has authorised the data and records
Manufacture of medicinal gases: Annex 6
Manufacture of active substance gases
Manufacturers are exempt from the PIC/S Guide to GMP requirements relating to bulk, liquified gas manufacture (Annex 6 clauses 1-3).
Training
All persons involved in the manufacture of medicinal gases should receive training relevant to their specific tasks and records of training should be maintained (Annex 6 clause 5).
All persons performing release for supply should be suitably trained and experienced in the manufacturing and test requirements of medicinal gases as well as the process of release for supply.
Separation of medicinal gases from non-medicinal gases
Manufacturers are required to implement controls to avoid mix-ups of products, which include separation of medicinal and non-medicinal gas production processes.
A ‘separate area’ for filling activities is usually achieved by filling medicinal gases on dedicated manifolds. Such an area should have sufficient space around to perform all the filling related tasks.
Storage of empty and filled cylinders
Gas cylinders should ideally be stored:
- under cover
- protected from the elements
- protected from potential contamination
Where justified by the manufacturer, it may be acceptable to store cylinders outdoors where the manufacturer implements controls including but not limited to:
- empty cylinders are checked to determine:
- the cylinder has a positive pressure to ensure that it is not emptied
- any faulty cylinders are quarantined and sent for further inspection and repair
- full cylinders have appropriate plugs and/or dust covers in place to protect the valve outlets
Prepare empty cylinders for filling under cover.
For filled cylinders, the level of cleanliness required in the storage area should be compatible with the expected environment in which they may be used by the customer.
Delivery of bulk gas into hospital tanks
Bulk liquified gas
Bulk liquified gas intended to be delivered into hospital tanks is exempt from:
- the operation of Part 3-3 of the Therapeutic Goods Act 1989 (Schedule 7 Item 17)
- Annex 6 clause 18 concerning records maintained for bulk gas intended to be delivered into hospital tanks, therefore does not apply to bulk liquified gas supplied in this manner
Bulk supply of non-liquified gases
Annex 6 clause 18 may apply to the bulk supply of non-liquified gases as the manufacture and supply of these gases is not exempt from the requirements of Good Manufacturing Practice.
Control of residual impurities
The stated maximum theoretical impurity level of 500 ppm v/v for a filling pressure of 200 bar at 15 °C (and equivalent for other filling pressures) (Annex 6 clause 32) will be understood to have been achieved by the required pre-fill cylinder preparation. Evidence that it is being achieved will be based on validation data, so routine testing will not be required.
Batch numbering of cylinders
Annex 6 requires that a batch be defined for filling operations (Annex 6 clause 31). Manufacturers should consider as a batch each group of cylinders filled:
- via a multi-cylinder manifold
- in an uninterrupted filling cycle, in case of cylinders filled one at a time
Numbering
The batch number:
- should be on each gas cylinder
- may be displayed on a separate label (Annex 6 clause 37)
- may be a validated unique identifier, such as an individual cylinder barcode label
Identification
Batch identification needs to:
- ensure traceability of a specific batch through all stages of manufacture and distribution (including all maintenance history)
- relate to each cylinder filled as part of the production batch manufacturing process
Traceability
To ensure adequate traceability of cylinders involved in a batch the following criteria concerning batch identification must be met:
- each filled cylinder must be traceable to significant aspects of the relevant filling operations
- the manufacturers cylinder control system must be able to identify all cylinders involved in a specific batch
- cylinders should be uniquely identifiable in human readable form to allow customers to identify and isolate cylinders involved in a specific batch in the event of recall action or a quality related incident
- The manufacturers’ cylinder control system must retain historic data for all batches a cylinder has been included in
Transportation of packaged gases
Filled gas cylinders should be protected during transportation (Annex 6 clause 45).
While Australian therapeutic goods regulations do not encompass good distribution practice (GDP) it is expected that the finished product will be released in a format that will provide adequate protection during transportation.
Where transportation of the packaged gases is likely to occur in open vehicles it is expected that cylinders will be fitted with appropriate plugs and/or dust covers to protect the valves, and such activities are recorded in the batch records.
Computerised systems: Annex 11
Implications of changes to Annex 11
Annex 11 has been updated to provide clarification of existing requirements to ensure that computerised systems are managed appropriately, particularly in relation to data management and integrity. In some cases, the wording of the clauses has become less prescriptive to allow better use of quality risk management principles in the validation and control of computerised systems.
Validation and control of computerised systems
Licenced manufacturers of medicines are to validate and control all computerised systems (including commercial off the shelf systems). This is in accordance with Annex 11 requirements (i.e. GMP computerised systems).
The level, extent and formality of system control should be commensurate with the criticality of the system. Manufacturers should have a good understanding of all the systems used, and the impact and criticality of each system.
In general, the following systems (list is not exhaustive) should be fully validated and controlled, such as those used:
- for the electronic acquisition of quality control data
- to control and monitor the operation of critical utilities, facilities and equipment
- to generate, store or access electronic GMP records
- to generate, process, calculate or monitor data that forms part of the batch processing record, or batch control testing records
- in place of physical (hard-copy) records, e.g. electronic spreadsheets used to track records or perform calculations, electronic documents used to record data
- to control the status of materials, products, equipment or processes, e.g. Enterprise Resource Planning systems
- to perform the release of materials and release for supply of finished goods
- to track the distribution of products and/or control the reconciliation of products and materials in the case of quality defects or recalls
'Regulated users' definition
The TGA regards 'regulated users' (Annex 11 clause 3.3) to be the licence or GMP certificate holder responsible for the application of GMP.
'Life-cycle' of a computerised system
The 'life-cycle' of a computerised system includes all stages from the initial concept, design, qualification, validation, and use through to the eventual retirement of the system and archival of all data.
Manufacturers need to manage computerised systems effectively at all stages in the life-cycle to ensure that they function correctly. Therefore, validation not only applies at the introduction of the system, but throughout all stages of use. Further guidance regarding the life-cycle management of computerised systems may be found within the PIC/S Good Practices for Computerised Systems in Regulated GXP Environments (available from PIC/S publications).
Qualification and validation (Annex 15)
All equipment used in the manufacture of medicinal products must be appropriately qualified. Follow the principles outlined in Annex 15 section 3. Acceptability of the approach taken will be assessed during inspections on a case-by-case basis.
Determine the nature and extent of qualification based on risk management principles. It is generally expected that all stages outlined in Annex 15 section 3 would be addressed in the qualification of new and/or complex equipment. Some of the stages may be omitted, where appropriately justified based on risk, depending on:
- use
- stage in the equipment life cycle
- nature of the equipment
Application of concurrent process validation
For medicinal gas manufacture, concurrent process validation is permitted.
Concurrent process validations should:
- be approved under the sites PQS
- make results and conclusion of any supporting data available to the authorised person performing release for supply
Number of batches used in process validation
Manufacturers are to determine and justify the number of batches used for process validation based on risk management principles. TGA’s general expectations are that:
- for a new process or product, a minimum of 3 batches are to be conducted for validation purposes
- for a process subject to technology transfer from one site to another, an extensive evaluation and risk assessment (with supporting data) should be in place to justify performing less than three batches. The assessment should cover the similarities and differences:
- in manufacturing processes
- equipment
- methods and materials
Performance qualification and process validation
For medicinal gas manufacture, performance qualification may be performed in conjunction with operational qualification and process validation.
Installation qualification and operational qualification can be combined as an installation/operation qualification.
Critical Quality Attributes (CQA) and Critical Process Parameters (CPP)
Importance and use of CQAs and CPPs
CQAs and CPPs:
- are important elements of product and process knowledge
- should be used in the design, validation and control of manufacturing processes
- can be used in periodic critical revalidation
Descriptions of CQA and CPP
CQAs:
- are physical, chemical, biological, or microbiological properties or characteristics that should be within an appropriate limit, range, or distribution to ensure the desired product quality
- are used to guide process development and control strategies
- can be modified as product knowledge and process understanding increase
- should have appropriate acceptance criteria to demonstrate product quality, including identity, assay, water content and any other relevant attributes
CPPs:
- are process parameters whose variability has an impact on CQAs
- should be monitored or controlled to ensure the process produces the desired quality
- should consider verification for process controls, such as evacuation of cylinders, fill pressure and weight
Ongoing Process Verification (OPV)
Periodically use ongoing process verification (OPV) to:
- evaluate process parameters and trends
- ensure that processes are consistent and remain in a validated state (Annex 15 clauses 5.28-5.32)
Use the outcomes from the OPV to look at any correlation between process capability and trends identified in the PQR. Base the frequency of OPV on risk management principles.
Investigate any adverse trends concerning product quality attributes to determine if processes are consistent and remain in a validated state.
OPV should be:
- used to support the validated status of the product
- documented in the Product Quality Review
Transport verification
The basic expectation is that manufacturers:
- will transport all products (including bulk products and finished products) in full accordance with appropriate storage conditions
- will not store or transport medicines outside their labelled and approved storage conditions
For medicinal products, clearly specify in quality or technical agreements responsibilities for:
- transportation
- monitoring
- storage
Further information about the transport of packaged medicinal gases is included in the Annex 6 clause 45.
Page history
Title changed from 'Medicinal gases guidance' to 'Understanding Good Manufacturing Practice (GMP) for registered medicinal gases' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updated to link to PIC/S Guide to GMP for medicinal products – version 16.
Change in title.
Addition of release for supply information.
Restructured and updated to be consistent with PE009-13, PIC/S Guide to GMP.
Original publication: Guide to interpretation of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products - 15 January 2009; applicable to the manufacture of medicinal gases
Replaced previous interpretation table on the TGA website for the Australian Code of GMP for Medicinal Products (16 August 2002).
Title changed from 'Medicinal gases guidance' to 'Understanding Good Manufacturing Practice (GMP) for registered medicinal gases' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updated to link to PIC/S Guide to GMP for medicinal products – version 16.
Change in title.
Addition of release for supply information.
Restructured and updated to be consistent with PE009-13, PIC/S Guide to GMP.
Original publication: Guide to interpretation of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products - 15 January 2009; applicable to the manufacture of medicinal gases
Replaced previous interpretation table on the TGA website for the Australian Code of GMP for Medicinal Products (16 August 2002).