Demonstrating the quality of listed probiotic medicines
Purpose
The purpose of these Guidelines is to help sponsors and manufacturers meet the technical, scientific and regulatory requirements to ensure the quality of their probiotic medicine is acceptable under the Therapeutic Goods Act 1989 (the Act).
Note: This document is a guide only
- These Guidelines are not mandatory requirements but rather show sponsors what a TGA delegate considers in a compliance review. However, there may be individual circumstances that justify a departure from these Guidelines and in any circumstance the TGA will consider the merits of each case against the regulatory requirements.
- Providing transparent and clear guidance on the elements considered by a TGA delegate in a compliance review is good regulatory practice and aims to mitigate the risk of non-compliance.
- It is the responsibility of each sponsor to understand and comply with the regulatory requirements contained in the Therapeutic Goods Act 1989 (the Act) and supporting regulations. You are encouraged to seek your own professional advice on how therapeutic goods legislation and other applicable laws apply to you.
Legislation
Scope
These Guidelines apply to probiotic medicines (with an AUST L or L(A) number) that are listed on the Australian Register of Therapeutic Goods (ARTG). Probiotics are defined as live microorganisms that when administered in adequate amounts, are proposed to confer a health benefit on the host.
Where these Guidelines provide interpretations of legislation, they are not mandatory requirements, but provide transparency to industry by showing sponsors what a TGA delegate considers when assessing the quality of listed and assessed listed probiotics in a compliance review. Additionally, these Guidelines are intended to assist with sponsor and manufacturer compliance by naming and explaining the most relevant legislation for ensuring the quality of probiotic medicines.
These Guidelines reference, state and summarise legislation and other guidelines that may be updated after these Guidelines are published. Sponsors are recommended to check the information herein against any updated and current versions.
These Guidelines do not apply to medicines with therapeutic activity attributed to distinct ingredients that are inactivated, non-viable microorganisms and/or their components (known as postbiotics or paraprobiotics). If a postbiotic is a distinct active ingredient within a probiotic medicine, then these Guidelines apply to the probiotic ingredients.
These Guidelines do not apply to probiotic foods[1] such as fermented dairy products, fermented teas and fermented vegetables.
The information in this guideline is complex, and careful reading is required to understand how it applies to a particular product. The complexity is due to the multiple regulatory requirements (section Applicable legislation) and then the multiple options for how compliance can be demonstrated (section Demonstrating compliance with legislative requirements). For example, for any particular product there are options for how to demonstrate quantification and options for labelling, and these depend on multiple factors: the product formulation, whether efficacy is at the strain or species level, and which default standard is applicable and chosen. As such, a topic covered in one section can depend upon the requirements and options explained in another section. For example, everything about labelling for a particular product will extend beyond the ‘Labelling’ section. Therefore, the reader’s pathway through this document will branch via references to other parts of the guideline.
Guideline structure
These Guidelines are divided into three main sections:
- Quality Control: outlines why it is important to control the quality parameters of probiotic medicines;
- Demonstrating compliance with legislative requirements: explains how to comply with the requirements outlined in Section Applicable legislation;
- Applicable legislation: details the legislation that sponsors must comply with to control the quality of their probiotic medicine.
Quality control
This section defines ‘quality’ and outlines why controlling quality parameters is important for the safety and efficacy of probiotic medicines and is thus important for the health and safety of Australians.
The control of quality parameters ensures that a listed medicine is consistently manufactured to result in a final product that meets the design specifications and is safe and efficacious throughout its shelf life.
Quality parameters
Quality is defined in subsection 3(1) of the Therapeutic Goods Act 1989 (the Act) as (bold added for relevance to the quality of probiotics):
- Quality, in relation to therapeutic goods, includes the composition, strength, potency, stability, sterility, purity, bioburden, design, construction and performance characteristics of the goods.
Figure 1 shows the quality parameters that relate to ensuring the safety and efficacy of probiotic medicines. There are legislative provisions in place that facilitate the control of these quality parameters, which is explained further in section Applicable legislation.
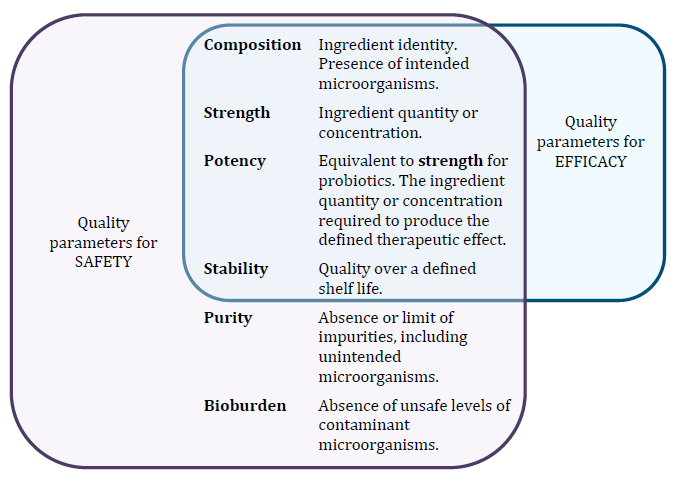
Safety
A listed medicine must be safe for the purposes for which it is to be used, and must not breach a condition of listing that may result in harm or injury (paragraphs 26A(2)(b) and 21A(5) of the Act, respectively).
The safety of listed medicines is partly controlled by only permitting the use of ingredients in the Therapeutic Goods (Permissible Ingredients) Determination (under Schedule 4, items 3 and 8 of the Therapeutic Goods Regulations 1990). Controlling the quality parameters (see Figure 1) of a probiotic medicine ensures that the probiotic medicine continues to be eligible for listing (i.e. contains an ingredient specified in the Permissible Ingredients Determination) and is suitable for supply as a low-risk medicine without TGA pre-market evaluation.
Bioburden
The definition of quality in subsection 3(1) of the Act includes bioburden, which is defined in the same subsection as:
- the quantity and characteristics of microorganisms present in the goods or to which the goods may be exposed in a manufacturing environment.
In the context of probiotic medicines, bioburden refers to the enumeration of contaminant microorganisms. The bioburden test must be established to have the ability to detect microbial contamination in the presence of the active probiotic ingredients (refer to sections titled Microbial contamination). The methods in TGO 100 for detecting microbial contamination are not suitable for probiotics (refer to Ministerial standards). Bioburden testing is conducted to ensure bioburden remains below acceptable limits in starting materials, product intermediates, final product and in ongoing stability studies throughout the product shelf life.
Sponsors must be able to demonstrate they have adequate control of bioburden in order for their medicine to be safe, and thus for it to remain on the ARTG. Sponsors are required to certify that the medicine is safe for its intended purpose (paragraphs 26A(2)(b)).
Efficacy
Efficacy is the capacity for therapeutic effect. The efficacy of a probiotic medicine depends on the presence of one or more specific therapeutically active ingredients in sufficient quantities over the shelf life of the medicine. As such, controlling the quality parameters in Figure 1 is critical for ensuring efficacy.
The ingredient in a probiotic medicine that is responsible for efficacy and stated in the information or evidence held by the sponsor (e.g. a clinical trial), is usually identified at the strain level; although, efficacy could potentially be attributed to a species or higher taxon. If a probiotic medicine’s efficacy is not strain specific (i.e. instead it is species specific), then any strain in that species could confer the same therapeutic effect, and thus any strain in that species could be an ingredient responsible for efficacy in the medicine. Similarly, if the medicine’s efficacy is genus specific, then any species (and strain within those species) could confer the same therapeutic effect and thus any species (and strain) in that genus could be an ingredient responsible for efficacy in the medicine.
How the quality parameters can be controlled is dependent on whether the efficacy of a probiotic medicine is genus, species or strain specific. This is further explained below, in section Demonstrating compliance with legislative requirements.
For more information about efficacy, indications and evidence, refer to the Supporting claims and indications for listed medicines, Therapeutic Goods (Permissible Indications) Determination and the Permitted indications for listed medicines guidance.
Stability
The stability of a medicine refers to a product that is shown to remain, or is likely to remain, within its design specifications (i.e. quality parameters in the product specification) throughout its shelf life when stored under the labelled storage conditions.
The quantities (and proportion) of active ingredients in probiotic medicines have the potential to vary from input at the start of manufacturing, to the final product at batch release, to the end of the medicine’s shelf life. This variability arises because the active ingredients in probiotic medicines are live microorganisms whose rates and extents of growth, and hence their viable quantities, are responsive to multiple factors. These factors include product formulation (available growth factors such as microbial metabolites, excipients, residual water and oxygen), manufacturing processes (stresses such as cryopreservation, lyophilisation and grinding), type of container closure, and storage temperature.
Variability in quantity and proportion also arises between different strains which can differ in their viability and growth due to their different genotypes and phenotypes. For example, different strains may have different capacities to maintain their viability in response to refrigeration, low oxygen availability and low residual moisture (water content or water activity).
As such, controlling the stability of a probiotic medicine is important for efficacy because throughout the shelf life of a probiotic, the quantity of an active microbial ingredient should remain no less than the claimed quantity on the label which provides the dose necessary for efficacy.
Controlling the stability of a probiotic medicine is also important for safety because throughout the shelf life of a probiotic, unsafe levels of specified contaminant microorganisms (bioburden) may occur.
Demonstrating compliance with legislative requirements
This section provides a practical summary of how sponsors can demonstrate compliance with applicable legislative requirements. The legislative requirements are explained in detail in section Applicable legislation.
Sponsors may demonstrate compliance using approaches that are different to those outlined in this section. However, sponsors will be expected to document justifications for how their alternative approaches achieve the legislated outcomes outlined in section Applicable legislation. An evidence-based justification of quality includes supportive high-quality scientific data (observations and measurements) that can be additionally strengthened by reasons or robust explanations using scientific concepts or mechanisms.
This section also discusses the TGA guidelines and international scientific guidelines such as the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), which suggest ways to demonstrate compliance with the legislation but are not intended to be restrictive and can enable innovation.
How to determine which quality standards to comply with
A quality standard refers to a default or ministerial standard. There may be multiple default standards (section Default standards) and ministerial standards (section Ministerial standards) that can apply to a listed probiotic medicine, but at least one default standard will apply.
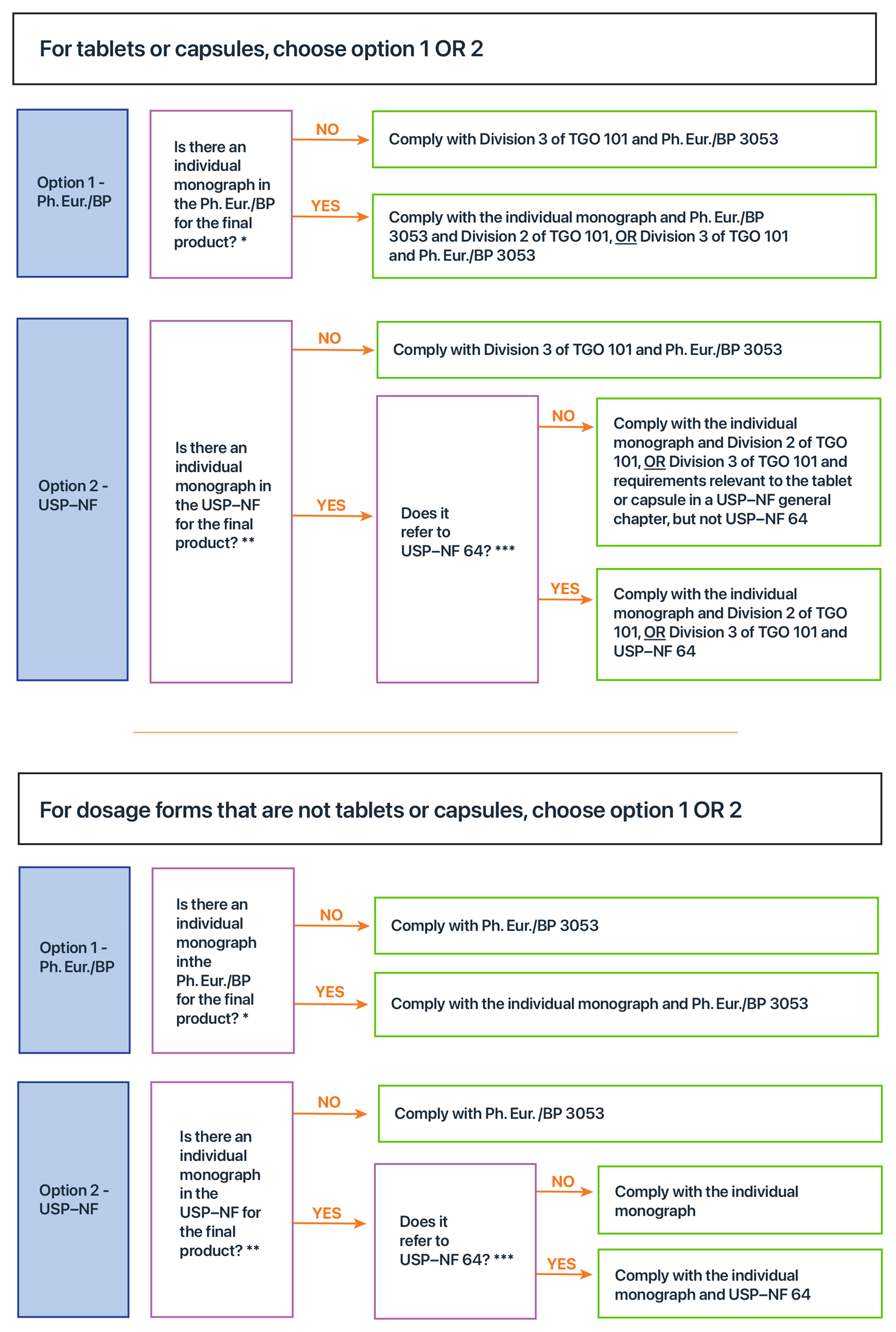
Figure 2 assists sponsors to determine which quality standards apply to their final product, depending on whether it is a tablet or a capsule, or some other dosage form. Figure 2 includes references to individual monographs (e.g. ‘Lactobacillus acidophilus’), general monographs (e.g. Ph. Eur./BP 3053), a general chapter (e.g. USP–NF 64) and a ministerial standard (TGO 101). To understand the rationale and details behind Figure 2 refer to sections Default standards and TGO 101 – Dosage forms that are tablets, capsules and pills.
Note: An individual monograph may apply even where the title of the monograph and the name of the medicine or ingredient are not identical. Where the name of a medicine or ingredient is a variant of the name of an individual monograph, the provisions of that monograph, including definitions, and the General Notices section of the relevant pharmacopoeia should be reviewed to determine whether the monograph applies.
Active ingredient identification, quantification and labelling
Microbial taxonomy
Microorganisms are named (taxonomically classified) by their genus, species (and sometimes subspecies) and strain. These classifications enable microorganisms to be identified as being different or similar to one another. The active microbial ingredients in a probiotic medicine have their identity, quantity and labelling at the taxonomic level of either strain (Genus species strain) or species (Genus species), including subspecies when relevant.
In these Guidelines:
- Strain-level taxonomy refers to naming that specifies the ‘Genus species strain’ e.g. Lactobacillus acidophilus La-14 or Bacillus coagulans (MTCC 5260).
- Species-level taxonomy refers to naming that specifies the ‘Genus species’ e.g. Lactobacillus acidophilus or Bifidobacterium animalis subsp lactis.
A strain is a population of microorganisms that descends from a single organism or pure culture isolate. Different strains of a single species differ in their sets of genes (genotype) and their observed characteristics (phenotype). The phenotype of a strain results from interactions between its genotype and the environment, which affects its biochemistry, physiology, developmental processes and behaviour.
Consequently, identification of active microbial ingredients in probiotics at the strain level ensures they are distinct from other microbial ingredients with potentially different characteristics, risks and therapeutic effects (indications). Generally, strain-level identification is important for all probiotics, whether they are single-strain or multi-strain products.
Many of the probiotic medicines that are currently listed on the ARTG contain several strains from at least one genus or species (see Table 1 for a potential example). These are called multi-strain probiotics.
| Genus | Species | Subspecies | Strain |
|---|---|---|---|
| Lactobacillus | delbrueckii | bulgaricus | (Lb-87) |
| acidophilus | (La-14) | ||
| Bifidobacterium | breve | (Bb-03) | |
| animalis | lactis
| (Bi-07) | |
lactis
| (Bl-04) |
Overview
The identity and quantity of an active microbial ingredient should be controlled and labelled at the appropriate taxonomic level. The taxonomic level for the active ingredient will depend on the:
- taxon responsible for efficacy in the evidence held by the sponsor
- taxon permitted for use in the Permissible Ingredients Determination
- taxon selected from the ARTG
- quality standards that apply to the product (see Figure 2)
- quality standards that apply to the starting material (see Figure 4).
Under the Act, quality testing for active ingredients must meet at least one default standard applicable to the medicine. European/British Pharmacopoeia monograph 3053 (Ph. Eur./BP 3053) is applicable to listed probiotic medicines. It requires identification and quantification of active ingredients at the strain level in a finished product. See section Compliance with default standards.
It is recognised that Ph. Eur./BP 3053 is not fully compatible with multi-strain probiotics and current industry testing capabilities.
The Ph. Eur./BP General Notices, which presides over all other sections of the Ph. Eur./BP, allows for quality of active ingredients to be controlled by alternative strategies (see section Alternative approach to compliance). This can include identification and quantification by input (QBI) which are widely used and when appropriately supported by evidence-based justifications, are considered equally acceptable. See sections Quality of starting materials and Quantified by input.
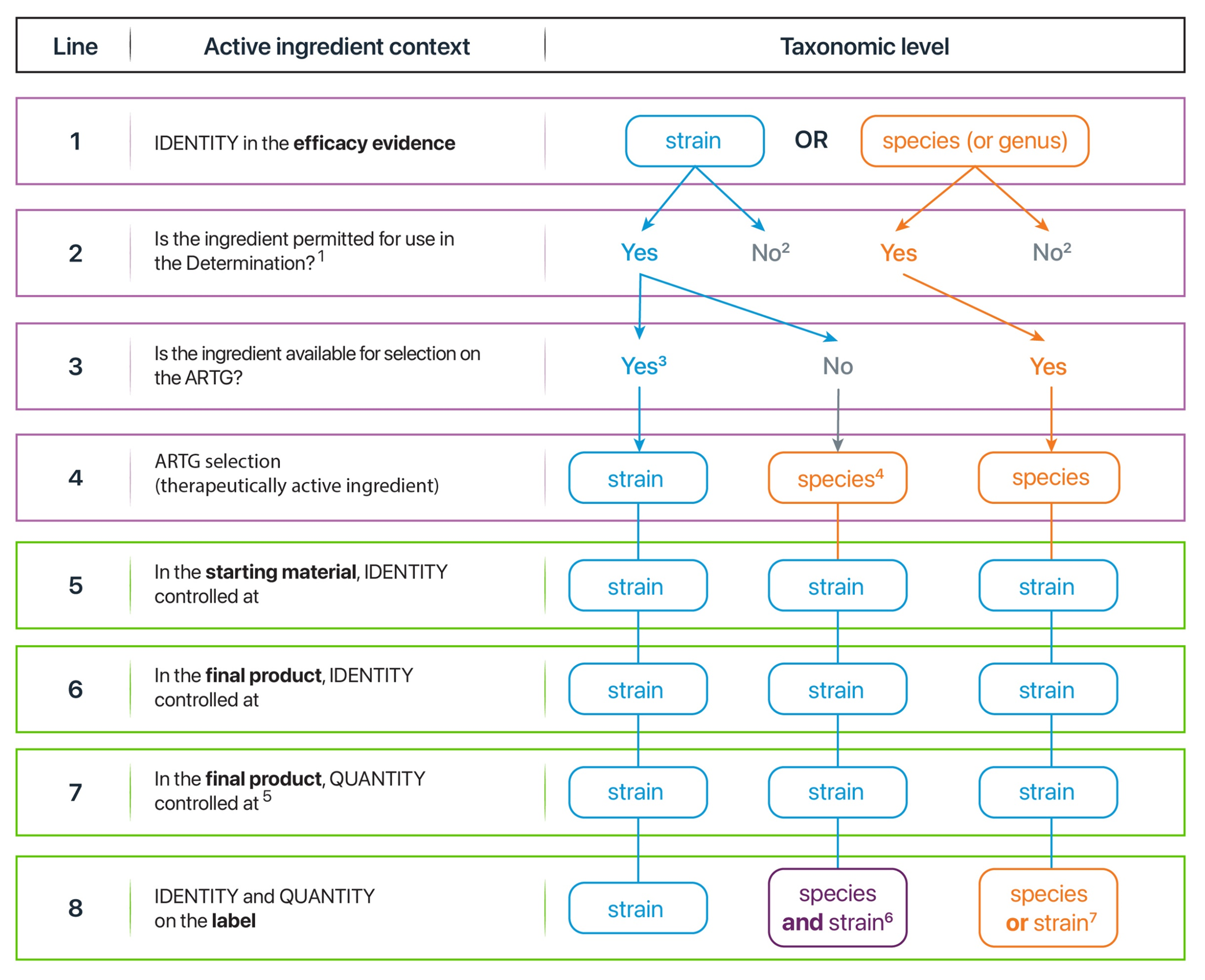
Figure 3 summarises the taxonomic level at which an active microbial ingredient is to be controlled and labelled. The footnotes to Figure 3 provide further explanation, and Table 2 refers to relevant explanatory sections in these Guidelines for each line in Figure 3.
Demonstrating that a quality parameter (e.g. strain quantity) has been sufficiently controlled can include using an evidence-based justification for how the required outcome is achieved through alternative strategies or a different test method.
Controlling the active ingredient does not always mean ‘testing’ for a particular quality parameter. For example, there are various ways the correct quantity (as labelled) of an active ingredient strain in a final product can be controlled other than testing (assaying) at strain level. Depending on the circumstance, Quantified by input (QBI) with an evidence-based justification and/or an assay at genus level may be sufficient to demonstrate that the quantity of a strain has been adequately controlled. Refer to the options in section Active ingredient quantification in the final product.
In Figure 3, rows 1 and 3 outline that the ingredient available for selection on the ARTG depends on the taxa that is responsible for efficacy. At the time these guidelines were prepared there are no strains available for selection on the ARTG.
Row 4 outlines that for strain-level efficacy, a strain is selected on the ARTG if its strain name is an ingredient in column 2 of the Determination OR its species name is selected if its species name is an ingredient in column 2 of the Determination, provided there are no specific requirements in column 4 preventing their use.
Rows 5-7 outline that the identity and quantity of the active ingredient is expected to be controlled at the strain level irrespective of whether efficacy is at the strain, species or genus level, and irrespective of whether the species or strain is claimed on the label (row 8). Controlling active microbial ingredients at the strain level ensures they are distinct from other microbial ingredients with potentially different characteristics, risks (such as antibiotic resistance, virulence factors) and therapeutic effects (indications). Strain-level expectations are also based on the default standards (Ph. Eur., BP and USP–NF).
The footnotes and references further explain that controlling the active ingredient identity and quantity at the strain level may be achieved through testing (in accordance with Ph. Eur./BP 3053 or with the USP–NF if an individual monograph applies), through alternative test methods (in accordance with Ph. Eur., BP and USP–NF) or through alternative strategies such as QBI if Ph. Eur./BP applies.
| Line | Guideline section |
|---|---|
| 1 | Efficacy Listing certification Conditions of listing Permissible ingredients Determination |
| 2 | |
| 3 | Permissible ingredients Determination |
| 4 | Permissible ingredients Determination |
| 5 | Quality of starting materials Ph. Eur. and BP for information about characterisation of starting materials. USP–NF for information about official substances, including starting materials. GMP – starting materials for information about identification of starting materials during manufacturing. |
| 6 | Active ingredient identification in the final product Active ingredient quantification in the final product The Act–Permissible Ingredients Determination for information about the Act and how it legislates the control of quality (e.g. identity and quantity). Ph. Eur. and BP and Table 6 for information about identification and quantification in the final lot. USP–NF and Table 8 for information about identification and quantification in the final lot when there is an applicable USP–NF monograph (e.g. single strain products) TGO 101 – Dosage forms that are tablets, capsules and pills for information about the stated content of an active microbial ingredient. |
| 7 | |
| 8 | How to determine which quality standards to comply with Labelling Ph. Eur. and BP and Table 6 for information about identification and quantification on the label. USP–NF and Table 8 for information about identification and quantification on the label when there is an applicable USP–NF monograph (e.g. single strain products). TGO 92 – Labelling |
Quality of starting materials
Figure 4 assists sponsors to determine which quality standards to comply with for their starting materials. If there is an applicable individual monograph in the European, British and US Pharmacopoeias, then the sponsor can choose which one to comply with. Once chosen, the sponsor should comply with the monograph in its entirety. Where there are no quality standards, sponsors are expected to develop the parameters and acceptance criteria to ensure the quality of their starting materials. For more details on how to determine whether an individual monograph applies, refer to section Compliance with default standards. For information on the GMP requirements for controlling the quality of starting materials, refer to section Starting materials.
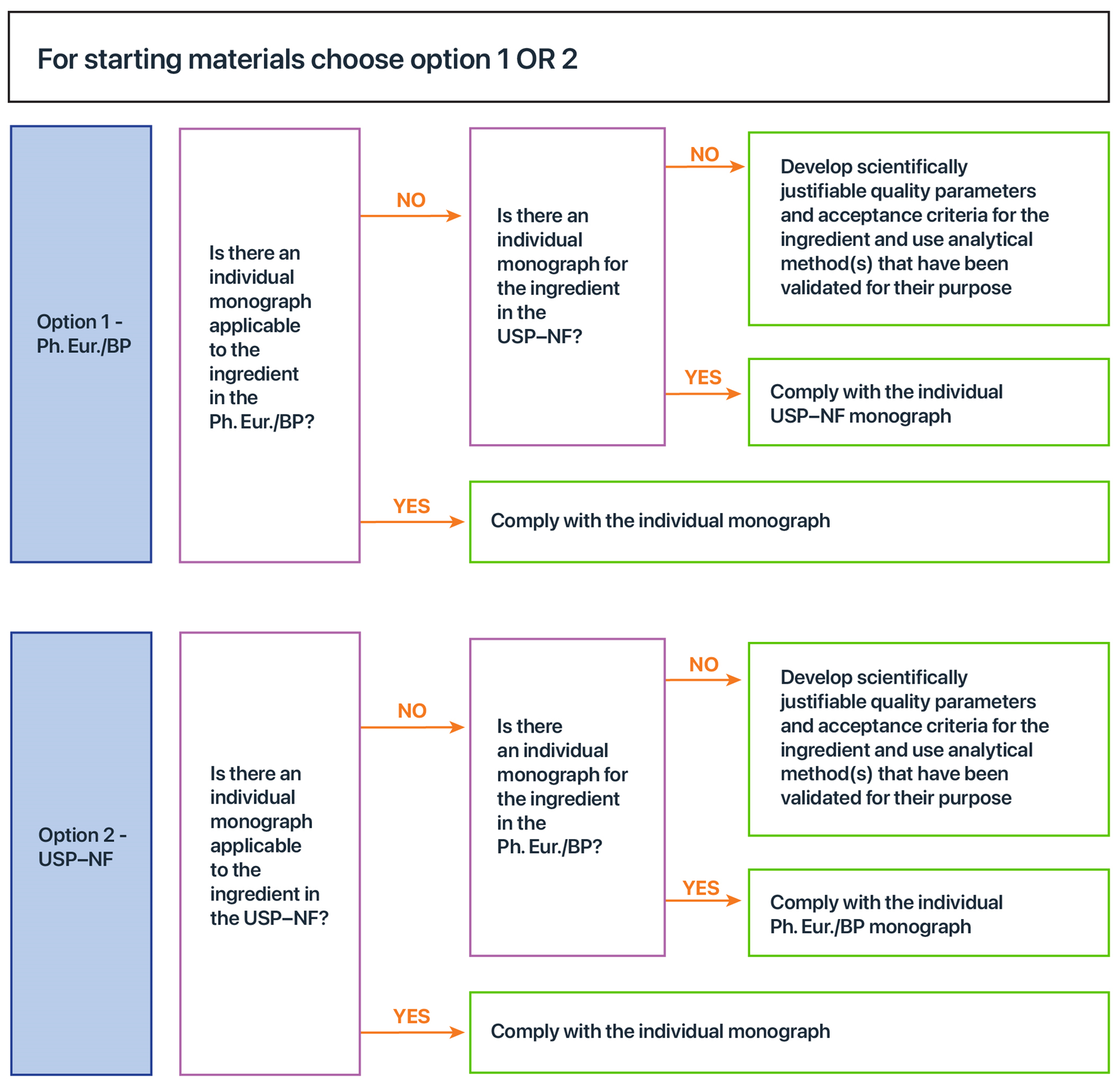
The requirements for starting materials, including testing of quality parameters, are explained in the following three guidelines. If a sponsor omits any of the steps outlined in these guidelines, then alternative measures to achieve the same level of quality assurance is necessary during later steps in the manufacturing process. For example, it may be possible for a sponsor or manufacturer to scientifically justify reduced testing of particular aspects of a starting material where extensive test history is established, which may justify conducting those tests on the final product instead. Conversely, if there is reduced testing of the final product (e.g. use of Quantified by Input), then alternative control measures, which could include comprehensive supplier qualification and/or sufficient testing of the starting material, may be used. However, sponsors will need to justify that these alternative control measures provide the same level of quality assurance as the level of quality that would have been achieved if the final product was tested. That is, sponsors would need to scientifically justify that by using their alternative control measures, at batch release the final product will contain the claimed amount of each viable active microbial ingredient strain (or species if applicable).
Key information from the relevant guidelines have been highlighted below. However, readers should read each guideline in full.
Sampling and testing for listed and complementary medicines
- For starting materials and intermediate products, there are different sampling requirements depending on whether the supplier has been approved and the individual material has been qualified or not.
- Until a supplier has been approved, and the starting material is qualified, each delivery of starting material should be approved by the Quality Unit only after full sampling and testing has been successfully undertaken.
- Once a starting material has been qualified, reduced sampling and testing can be performed; as outlined in the sampling and testing Guidance referenced above. A manufacturer must perform critical tests on each delivery and is permitted to rotate through all non-critical tests. An assay is critical if it is part of release specifications. It is not permissible to skip any test without adequate justification. Critical tests and non-critical tests must be defined and a suitable sampling plan adopted.
- Identification is considered a critical test for all starting materials. The TGA guideline PE009, the PIC/S guide to GMP for medicinal products – TGA interpretation and expectations for demonstrating compliance explains that a discriminatory identification test should be used to correctly identify the strain or strains of organisms claimed on the product label. Biochemical identification methods may not be reliable for this purpose and genotypic identification methods should be employed, for example, 16S rRNA sequencing or PCR.
- Quantification, of a probiotic with appropriate acceptance limits, is also considered a critical test where a starting material label claim is made for the active ingredient.
- Other tests may also be critical, depending on the material and its functionality in the product, particularly where the material attributes are critical to ensuring product quality and/or stability. For example, a starting material should be tested to ensure that the ingredient complies with specific requirements in the Permissible Ingredients Determination, or where moisture (water activity) is a parameter that is critical to the stability of the probiotic good.
- For intermediate and bulk products, verify that each delivery is from an approved manufacturer with a current TGA licence or GMP clearance (PIC/S Guide to GMP, Part 1, Chapter 1). This usually includes receipt of the manufacturer’s Certificate of Analysis (C of A). Conduct sufficient testing to ensure the quality of the product, particularly where the quality may have been impacted during transport.
Starting material analytical procedure validation for complementary medicines
- Test procedures for starting materials, including quantification and identification of the active substance, must use analytical methods that are validated for that purpose.
- If the analytical procedure for starting material testing is included in the current edition of the Ph. Eur., BP or USP–NF, then validation of that procedure is not required. However, method transfer should be undertaken to ensure the test method is applicable at the point of use and appropriate for its intended purpose.
- A formal GMP or technical agreement for suppliers and manufacturers of raw materials is not required provided there are approved specifications, a C of A from the manufacturing site and appropriate records of supplier approval.
- There must be an approved specification (generated by the manufacturer) and C of A (from the supplier) available, including assurance of compliance with pharmacopoeial standards (e.g. Ph. Eur. 3053), such as strain characterisation. Characterisation of probiotic starting material includes strain-level identification and should include critical safety information such as antimicrobial resistance and susceptibility profile, the absence of virulence factors, and toxigenic and pathogenic attributes. Documentation of strain characterisation should be held by the sponsor and manufacturer.
- All suppliers should be approved before materials and products are used. Approval allows an assessment to be made of the suitability of the supplier to consistently provide the starting and/or packaging material to the manufacturer of a medicinal product. No materials should be released or used before the satisfactory completion of evaluation by the Quality Unit unless there are appropriate systems in place to allow for such use.
- Until material received from an approved supplier is qualified, each delivery of starting material should be approved by the Quality Unit only after full sampling and testing has been successfully undertaken.
- Reduced sampling and testing of starting materials may be performed once the supplier has been qualified and other conditions are met.
Active ingredient identification in the final product
Sponsors should demonstrate that the active ingredients identified on the label are in the final product. For legislation applicable to identification testing and quality control, refer to sections Default standards and Good manufacturing practice in these Guidelines.
Sponsors should identify the active ingredients in the final product at strain level, which is generally irrespective of whether the taxon responsible for efficacy is at the genus, species or strain level. Refer to Figure 3, Table 6, Table 8 and section Default standards.
However, if the taxon responsible for efficacy in their efficacy evidence is at species or genus level, then sponsors should check the current default standard (Ph. Eur., BP or USP–NF) that is relevant to their product to assess whether the standard has changed since publishing this guideline such that identification may be at species or genus level instead of strain level.
Ph. Eur./BP 3053 requires that each microorganism in the final lot must be identified at the strain level by a suitable method. A suitable identification method is one that has been validated for the intended purpose and has thereby demonstrated its ability to unequivocally identify the microorganism in the product at the level of strain.
A relevant USP–NF individual monograph will require an identification test be undertaken and that all requirements of all specified procedures in the test, including acceptance criteria, must be met to satisfy the requirements of the test.[2] The individual monographs and identification methods are currently specific to one or more strains. Refer to USP–NF 64 if cited in the individual monograph for identification purposes. USP–NF 64 explains that probiotic microorganisms are typically identified at the strain level as their characteristics are usually strain specific. USP–NF 64 provides a general testing procedure for the identification of Lactobacillus and Bifidobacterium strains by PCR with specific primers; although it refers to using the specific testing procedure, primer set and acceptance criteria in the relevant individual monograph.
If the Ph. Eur./BP is applicable and a sponsor chooses to use the Ph. Eur./BP (see section How to determine which quality standards to comply with), the identity of the strain can be controlled through alternative means (i.e. other than testing the final lot). Where scientifically justified, the sponsor may control the identity of the active ingredients in the final product through appropriate controls of the starting material and through comprehensive supplier qualification, such as those outlined in section Quality of starting materials. However, it is the sponsor’s responsibility to scientifically justify and ensure that the identity of the active ingredients remain unaffected by downstream processes and meet product specifications and label claims, despite not testing the final lot.
If the USP is applicable and a sponsor chooses to use the USP (see section How to determine which quality standards to comply with), then alternative means to achieve the active ingredient identity requirements set in the USP individual monograph and USP 64 would not be permitted (unless alternative means are specifically allowed for that purpose in the USP individual monograph).
Active ingredient quantification in the final product
Sponsors should demonstrate that the active ingredients identified on the label are in the final product at the correct quantities (i.e. as stated on the label).
Strategies to control the quantity of the active ingredient at strain level can include Quantified by Input (QBI) when combined with other supporting evidence such as (but are not limited to):
- supplier qualification to ensure that the starting material is indeed the strain that is intended to be in the probiotic
- blending process validation to ensure that the proportions of each strain that end up in each capsule are correct
- contamination testing to ensure that unintended microorganisms are not present and cannot take over
- water activity testing to ensure that the active ingredient quantities stay within the specifications throughout shelf life.
Sponsors can decide what strategies to implement to control the quantity of the active ingredient at strain level. Options include but are not limited to:
- testing the final product at strain level by using pharmacopoeial methods that have been verified for their intended purpose (sections How to determine which quality standards to comply with and Default standards) or by using validated alternative methods (various methods are outlined in section Selecting and validating quantification methods),
- scientifically justifying at strain level by testing the final product at species or genus level depending on the formulation (scenarios 1–3 Figure 5),
- using quantified by input (QBI) with an evidence-based justification (section Quantified by input), or
- demonstrating in some other way that is scientifically justified.
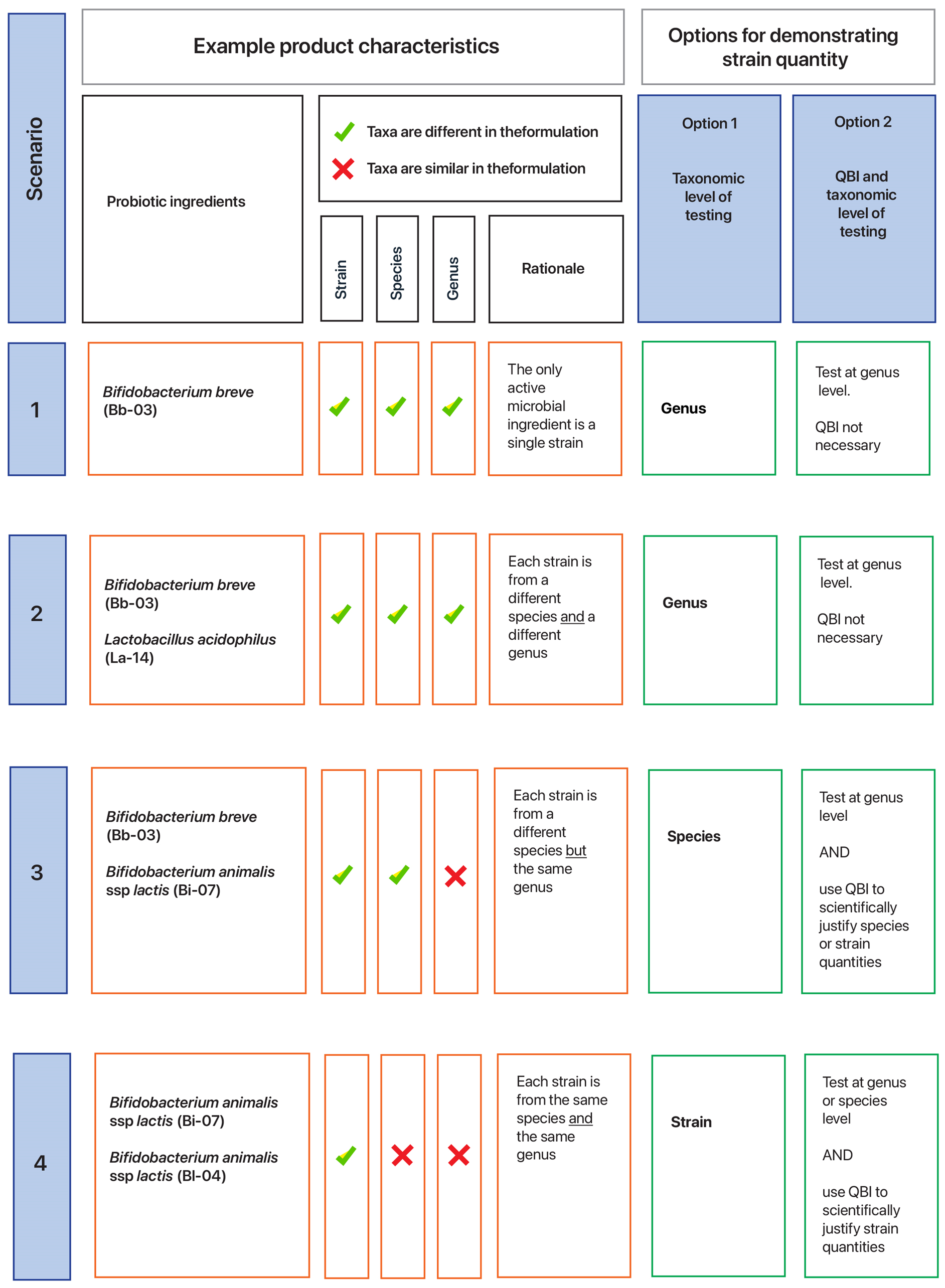
One strategy is to conduct strain-level quantification as part of batch release testing (refer to Ph. Eur./ BP 3053 in section Ph. Eur. and BP and USP–NF monographs and chapters outlined in section USP–NF). The quantification test method chosen depends on the blend of ingredient taxa in the medicine formulation, and thus whether strains can be quantified by testing the final product at the level of either strain, species or genus (Figure 5).
For example, a test at genus level (e.g. plate count method) could provide strain quantification data if there is only one strain per genus in a probiotic with multiple active ingredients (Figure 5, scenario 2) or if the probiotic only contains one active microbial ingredient that is a single strain (scenario 1). However, if a medicine contains more than one strain per species then a strain-specific method to measure either CFU or viable cells is required (scenario 3).
Strains cannot be quantified at the level of genus or species in scenario 3, or genus in scenario 2 as the test result cannot distinguish the quantities of the different strains. Furthermore, such test results cannot detect any change in the ratios of the strains in the final product at batch release or at the end of the medicine’s shelf life.
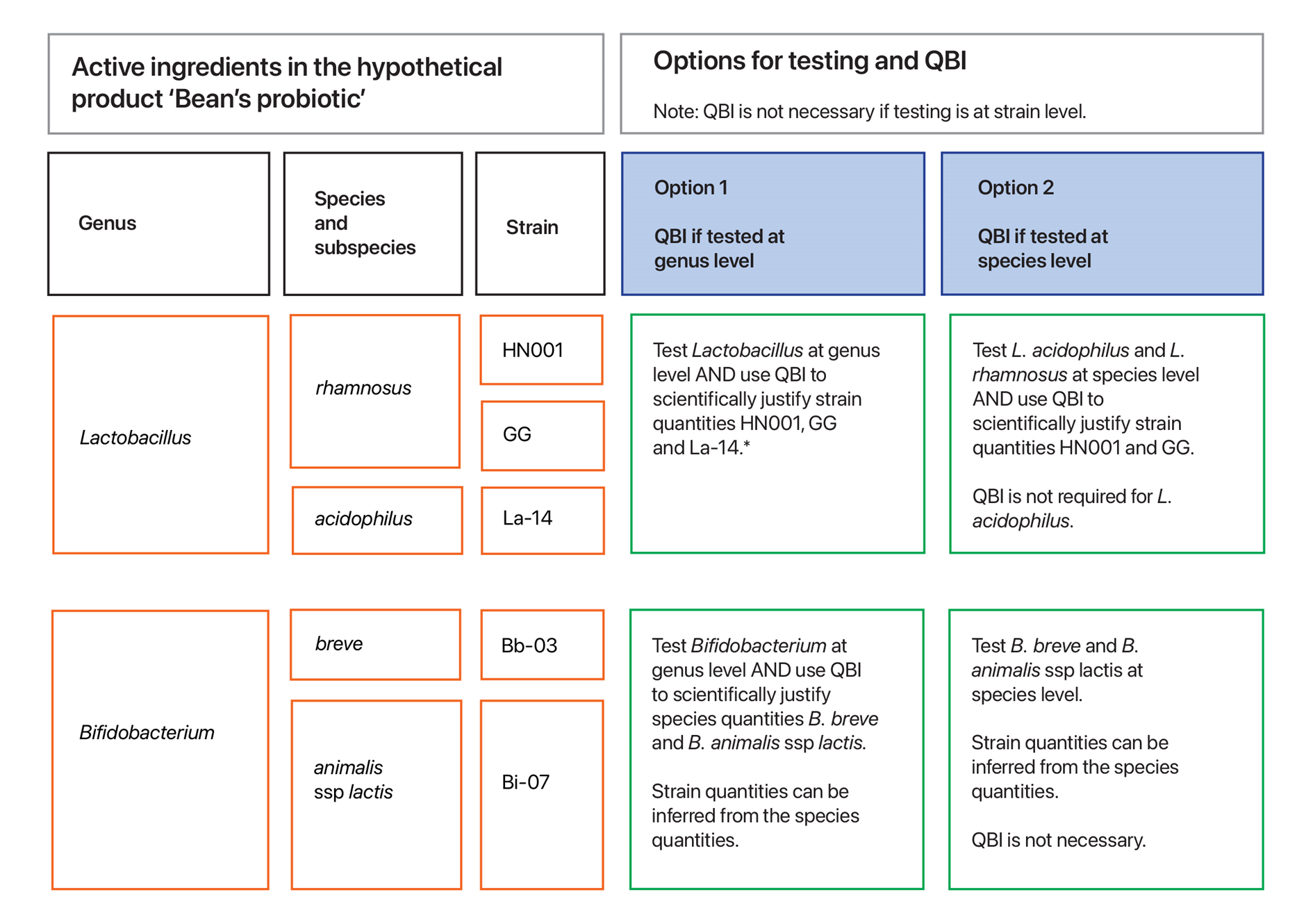
If the Ph. Eur./BP is applicable and a sponsor chooses to use the Ph. Eur./BP (see section How to determine which quality standards to comply with), an alternative strategy for enumerating to strain level in the final product is QBI (option 2 in Figure 5, and refer to section Quantified by input). QBI may be appropriate when the data and information to scientifically justify this approach is held by the sponsor and manufacturer and it demonstrates that the quantity of each strain in the final product as stated on the label is accurate.
Sponsors should choose a strategy most appropriate for their circumstances. For example, if access to validated strain quantification methods is not available, then whichever alternative strategy is selected, sponsors must still be able to demonstrate that the correct amounts of active ingredients, as stated on the label, are in the final product.
If the USP is applicable and a sponsor chooses to use the USP (see section How to determine which quality standards to comply with), then alternative strategies (such as use of QBI) to achieve the active ingredient quantification requirements set in the USP individual monograph and USP 64 would not be permitted (unless alternative strategies are specifically allowed for that purpose in the USP individual monograph). Alternative strategies for active ingredient quantification is not currently permitted because the available USP individual monographs (at the time this guideline was published) either require a specific enumeration method to be used, or they refer to Probiotic Tests <64> Enumeration for non-spore-forming bacteria strains which only allows an alternative validated microbiological procedure for quantification to be used if its equivalence to the pharmacopoeial procedure has been demonstrated. Additionally, at the time this guideline was published, there are no USP individual monographs for multi-strain products.
Selecting and validating quantification methods
Analytical procedures (i.e. test methods or assays) for quantifying the active ingredients in a medicine must be validated (refer to section GMP – Testing for quality). The selected procedures should depend on whether a medicine has either one strain or multiple strains per genus or per species (see Figure 5 above for examples). Useful information can be found in the TGA guideline Finished product (medicine) analytical procedure validations for complementary medicines and the ICH guideline Q2(R1) Validation of analytical procedures: Text and methodology. ICH Q2(R1) refers to characteristics for consideration during validation to ensure the analytical procedures are suitable for their intended purpose and provides a collection of terms and their definitions.
Culture methods are useful for quantifying microorganisms at the level of genus, but it is generally difficult or impossible to distinguish microorganisms at the strain level using these methods. Although, some species within the same genus can be differentiated using selective and differential culture media under different incubation conditions but the method should be validated for such a purpose. Culture methods are also limited as they cannot detect viable but non-culturable cells. Nevertheless, if this is the method selected, sponsors still need to demonstrate that each active ingredient is in the final product at the correct amount to support efficacy. There are various ways sponsors can do this, including QBI. Refer to sections Active ingredient quantification in the final product and Quantified by input.
Other methods that are currently being researched (e.g. nucleic acid, fluorescent dye, cellular and metabolic property -based methods) may in future be capable of quantifying viable multi-strain probiotics to strain level. Where a sponsor chooses to use an alternative analytical test or procedure to those presented in the relevant default standards, the test must be appropriately validated to demonstrate that it is suitable for its intended purpose (see section Alternative tests for identification and quantification).
Assay limit for content
Sponsors and manufacturers should consider the statistical variability in their quantification data during manufacture and method validation so that the quantity of each active ingredient in the final product is no less than the stated content throughout the shelf life of the product.
The stated content is the quantity stated on the label and claimed to be present in each tablet, capsule or other dosage form, and is the same as the quantity documented in the final product specification. For example, ‘Lactobacillus acidophilus, no less than 2 billion CFU/capsule’.
GMP requires specifications for final products to include or provide reference to the quantitative requirements (i.e. the expected final yield) with the acceptance limits (PIC/S Guide to GMP for Medicinal Products - Part I, Chapter 4).
For dosage forms that are tablets or capsules (and when following the Australian specific requirements in Division 3 of TGO 101), ensure that the assay limit for the content of each active microbial ingredient in the final product is ‘not less than stated content’ (subsection 14(2) of Division 3 and item 4 of Schedule 2 of TGO 101). Stated content is defined in subsection 4(1) of TGO 101 as the quantity of the active ingredient that is stated on the label and is thus claimed to be present in each capsule (or tablet). Refer to section TGO 101 – Dosage forms that are tablets, capsules and pills of these Guidelines.
For dosage forms that are not tablets or capsules, a relevant default standard applies instead of TGO 101. The statements in monographs from a relevant default standard (e.g. statements about an assay limit for content) are interpreted in accordance with the general notices from that default standard. The general notices of the Ph. Eur. and BP (Section 1.5.1.9) and the USP–NF (Section 4.10.20) require that a limit or acceptance criteria includes or allows for analytical error and no further tolerances are to be applied to the limit. This means that the quantity of each strain in the final product targets the quantity on the product specification and as labelled, but the measured quantity (on the certificate of analysis for each batch) must be no less than (NLT) the lower assay limit stated in the product specification. For example, Ph. Eur./ BP 3053 requires the potency of each strain to be not less than the stated value or it is within the stated range. An example from the USP–NF is the individual monograph for Lactobacillus acidophilus which applies an enumeration acceptance criterion of NLT 100% of the labelled viable cell count, in CFU/g, of the specified strain of Lactobacillus acidophilus.
Consequently, for the final product to comply with the assay limit for content, the following should be accounted for when determining the amount of overage required at the start of manufacturing:
- measurement error (statistical variability) for the product-specific assay,
- the quantity losses throughout the shelf life of the medicine and
- the claimed quantity for efficacy on the medicine label.
Accounting for these factors should ensure that at the end of the product’s shelf life, the product can be demonstrated to contain no less than the stated content (refer to section Overage).
Quantified by input
Quantified by input (QBI) is a practice used to estimate the content of an active ingredient in a final product (at batch release and stated on the finished product C of A) when it is not possible or practical to conduct an assay. QBI may be justified when a validated assay method for the ingredient in a final product is unavailable or is difficult to achieve because the medicine formulation is complex, or if an assay shows unresolvable matrix interference. Refer to the Australian regulatory guidance Quality for listed medicines for general QBI information.
It cannot be assumed that the quantities and ratios of individual viable strains in a final probiotic product are the same as the quantities and ratios at input as raw materials. Nor can it be assumed that any particular strain will grow or survive and remain viable in the same way as another strain or their species or genus (refer to section Stability).
QBI under Ph. Eur. and BP
Although Ph. Eur./BP 3053 requires strain quantities to be controlled in the final product, this does not mean that the final product always need to be ‘tested’ at strain level. The use of QBI supported by evidence-based justifications will be considered to meet the requirements of Ph. Eur./BP 3053. Refer to section Alternative approach to compliance.
The quantity of individual viable strains in a medicine can be determined by assaying at genus, species or strain level depending on the product formulation. Assays at genus level (and sometimes species level) using the plate count method are usually possible and practical, therefore QBI is not usually justified when genus (or species) level assays can demonstrate the quantity of each strain in the medicine. For examples, refer to scenarios 1, 2 and 3 in Figure 5.
However, QBI is an option when assays at genus (or species level) are insufficient to demonstrate the quantity of each strain in the medicine, and when a strain-level assay is not possible or practical (refer to Figure 5, scenario 3, when a genus-level test is insufficient) and scenario 4, when a species-level test is insufficient).
If a sponsor chooses to use QBI, an evidence-based justification is required to demonstrate that at batch release the final product will contain the claimed amount (‘no less than the stated content’) of each viable active microbial ingredient strain (or species if applicable; refer to Figure 3). Although, by design, the overage in the final product should ensure the product exceeds the claimed amount beyond batch release and through to the end of the product’s shelf life (refer to section Overage).
An evidence-based justification for how QBI is able to control the active ingredient at strain level can include multiple sources, for example:
- Raw material supplier qualification and strain-level identification and quantification of the raw microbial materials. Refer to section Quality of starting materials.
- Process validation data (to demonstrate uniform composition of raw ingredients in product dosage units) to support a claim that the amount of added starting material (of each probiotic ingredient) is the same as the amount in the final product. For example, mixing validation studies using surrogate analytes to represent different probiotic strains could be performed to demonstrate that appropriate quantities of each ingredient are present in the in-process bulk substance. Also, filling validation studies using surrogate analytes to represent different probiotic strains could be performed to provide assurance that the appropriate quantity of each probiotic is present in each dosage unit. Where a product grouping approach is used for process validation, document the scientific justification of the rationale used to establish product groupings.
- Data or information to demonstrate that during manufacture the viable quantity of one of the component strains does not decrease to below the label claim while concurrently others in the same species increase to compensate. Such strain-level changes are not detected when assaying for total genus or total species. Evidence could include water activity data.
Water activity data collected during product development, process validation, and/or routine testing of the final product to demonstrate how water activity is relevant to strain dormancy. For example, scientific data to substantiate a claim that when the water activity is below a certain value the change in quantity of a specific strain or a mixture of strains will be minimised over the shelf-life period. For the data to be relevant to the product, the water activity test should be conducted on a sample comparable to the product in relation to ingredient components, the type of container, and the storage temperature and humidity.
If controlling water activity forms part of a sponsor’s strategy to ensure the quantity of active ingredients in a final product, then pharmacopoeial methods should be used. Pharmacopoeial methods include USP–NF <922> ‘Water activity’ (which includes an application for optimising the stability of probiotics); Ph. Eur./BP 3053 includes water activity (2.9.39); and BP Appendix IX M ‘Water-solid interactions: Determination of sorption-desorption isotherms and of water activity’.
Figure 6 shows a hypothetical example of how quantification of each ingredient in a multi-strain probiotic can be supported by using QBI. Refer to Figure 5 for further rationale.
QBI under USP–NF
The USP–NF does not allow alternative control measures (such as QBI) to replace a specified test. The applicable individual monograph[4] will outline the specific tests required for a final product. The only alternative approach USP–NF allows is the use of an ‘alternative method or procedure’ that has been fully validated and produces comparable results to the compendial[5] method or procedure within allowable limits. Refer to section Alternative tests for identification and quantification. For example, flow cytometry is an alternative non-culture-based quantification method that has enhanced precision and is better suited to automation than the compendial method.
Stability of the final product
Stability studies are used to assure that a medicine’s specifications (such as active ingredient identity and quantity) will be maintained under the conditions described on the medicine label for the duration of its shelf life. The shelf life of a medicine should be based on scientific data.
At the time of listing the product on the ARTG, sponsors are expected to hold initial stability data[6] to support their certification that the medicine’s specifications will be maintained under the conditions set out on the medicine’s label until the medicine’s expiry date (paragraph 26A(2)(fc) of the Act).
After listing, stability data is required upon release of the product to support its shelf life and to confirm that the batch meets the requirements of the marketing authorisation. The TGA guideline on the PIC/S guide to GMP for medicinal products recommends basing on‑going stability studies on the principles of ICH Q1 Stability. The ICH guideline ICH Q1A(R2) Stability testing of new drug substances and products refers to using analytical procedures that are fully validated and stability indicating. The TGA Technical guidance on the interpretation of the PIC/S Guide to GMP On-going stability testing for listed and complementary medicines describes ways that a manufacturer may operate to demonstrate compliance with the relevant manufacturing principles.
An example of a stability indicating test that is relevant to probiotics is ‘water activity’. When water activity is within the limit or range on the product specification, the change in quantity of a specific strain or mixture of strains due to water can be minimised over the shelf-life period. Refer to section Quantified by input for a list of the pharmacopoeial methods.
If strain quantities in the final product have been controlled using QBI, then stability testing should be conducted at least at the same taxonomic level as used for the final product and an evidence-based justification is required to demonstrate that the claimed amount (‘no less than the stated content’) of each viable active microbial ingredient strain (or species if applicable; refer to Figure 3) is maintained in the final product for the duration of its shelf life. Refer to sections Active ingredient quantification in the final product, Quantified by input, and option 2 in Figure 5.
An evidence-based justification for stability may include the following:
- Stability data for active ingredients could be generated for individual strains, or combinations of strains, in conjunction with any other relevant information. Consideration should be given to whether other ingredients in the products formulation may affect the quantity of the probiotic over time. This type of data should demonstrate that the stability of each strain is not significantly affected when the strains are combined in a product under the claimed storage conditions and throughout its shelf life.
- The above stability data could include water activity data to substantiate a claim that when the water activity is below a certain value, changes in the quantity of a specific strain or a mixture of strains due to water will be minimised over the shelf-life period. For the data to be relevant to the product, the water activity test should be conducted on a sample comparable to the product in relation to ingredient components, the type of container, and the storage temperature and humidity.
- Stability data from a factorial design study (such as to assess the effect of strain combinations on each strain) may be applicable (refer to ICH Q1D, bracketing and matrixing designs for stability testing of new drug substances and products). For example, test samples are created with Lactobacillus acidophilus (NCFM) and two other strains so that all samples contain all possible combinations of one, two or three strains. All samples are then tested under conditions relevant to the product(s), including but not limited to storage time, temperature, humidity, active ingredients, excipients and container type. Then, if the number of viable cells of L. acidophilus (NCFM) remains stable in all samples for the duration of the test, then this data could demonstrate that the stability of L. acidophilus (NCFM) is not significantly affected when in a product containing any of the two other strains and under similar conditions.
Accelerated stability studies
Accelerated stability studies are unlikely to substantiate the stability of a probiotic medicine. Microbial viability (growth and decay) responds to multiple factors, and data from earlier time points in a stability study (such as at 6 months) may not reflect any clear trend towards a later endpoint. Therefore, unless demonstrated or scientifically justified, the viability of microorganisms in an accelerated study of a final product stored at 25°C for 6 months, as an example, cannot be assumed to be the same as their viability in the final product stored at 2–8°C for 24 months.
Any extrapolation from an accelerated or a short-term study to the full-term shelf life of a probiotic medicine should be supported by scientific data to demonstrate that the mathematical model or equation used in the extrapolation is reliable and valid.
Grouping of on-going stability testing
A listed medicine may be grouped into an on-going stability program for a representative medicine instead of being placed on its own specific on-going stability program. Further information can be found in the TGA guideline On-going stability testing for listed and complementary medicines.
The grouping approach should be supported by scientific data demonstrating that the formulation (all ingredients), dosage form, method of manufacture and primary packaging material are comparable between the listed medicine and the representative medicine. Additionally, grouped probiotic medicines should also be supported by scientific data demonstrating that the stability of any different genera, species or strains in the medicine are comparable to the reference medicine on the stability program. Any differences in the rates of change of strain quantities over time between the historical data and real-time data should be evaluated scientifically to assess whether the data supports the stability of the product.
The active ingredients in probiotic medicines are live microorganisms whose rates and extents of growth, and hence their quantities and viability, are responsive to multiple factors (refer to section Stability). Therefore, adequate data about stability-indicating variables (e.g. viable quantities) of each active ingredient strain should be collected to demonstrate that grouping with a representative medicine is appropriate.
Overage
Overage is the quantity of an ingredient added at the start of manufacturing that is additional to the claimed quantity. The purpose of overage is for a product to maintain the claimed amount of viable active microbial ingredient despite any known decreases throughout manufacturing and the product’s shelf life.
The overage for each active microbial ingredient should be considered during manufacture and during analytical method validation so that the quantity is no less than the stated content throughout the shelf life of a product (refer to section Assay limit for content).
The amount of overage should also be considered when a sponsor certifies their product is of acceptable safety (paragraphs 26A(2)(b) and 26A(2)(f) of the Act).
Labelling
If an active microbial ingredient name is at the level of species (‘Genus species’) in the Australian Approved Name (AAN) list and in the ARTG entry for the medicine, then the species name must be on the label of that medicine to comply with TGO 92, paragraph 8(1)(b). Refer to the Approved Biological Names (ABNs) list, which is a category within the AAN list.
If Ph. Eur./BP 3053 is an applicable standard, then in addition to the species name, the strain name (‘Genus species strain’) must also be on the label (Table 6). Alternatively, if the applicable standard is an individual monograph in the USP–NF, then follow the recommendation for labelling in that monograph (section USP–NF) and in USP–NF 64 if cited in that monograph (Table 8). If following the USP–NF, the identity and quantity on the label could be required to be at the strain level or at the species level.
Refer to Figure 3 (line 8) for a schematic showing when the name of an ingredient is required to be on the label at strain or species level or both.
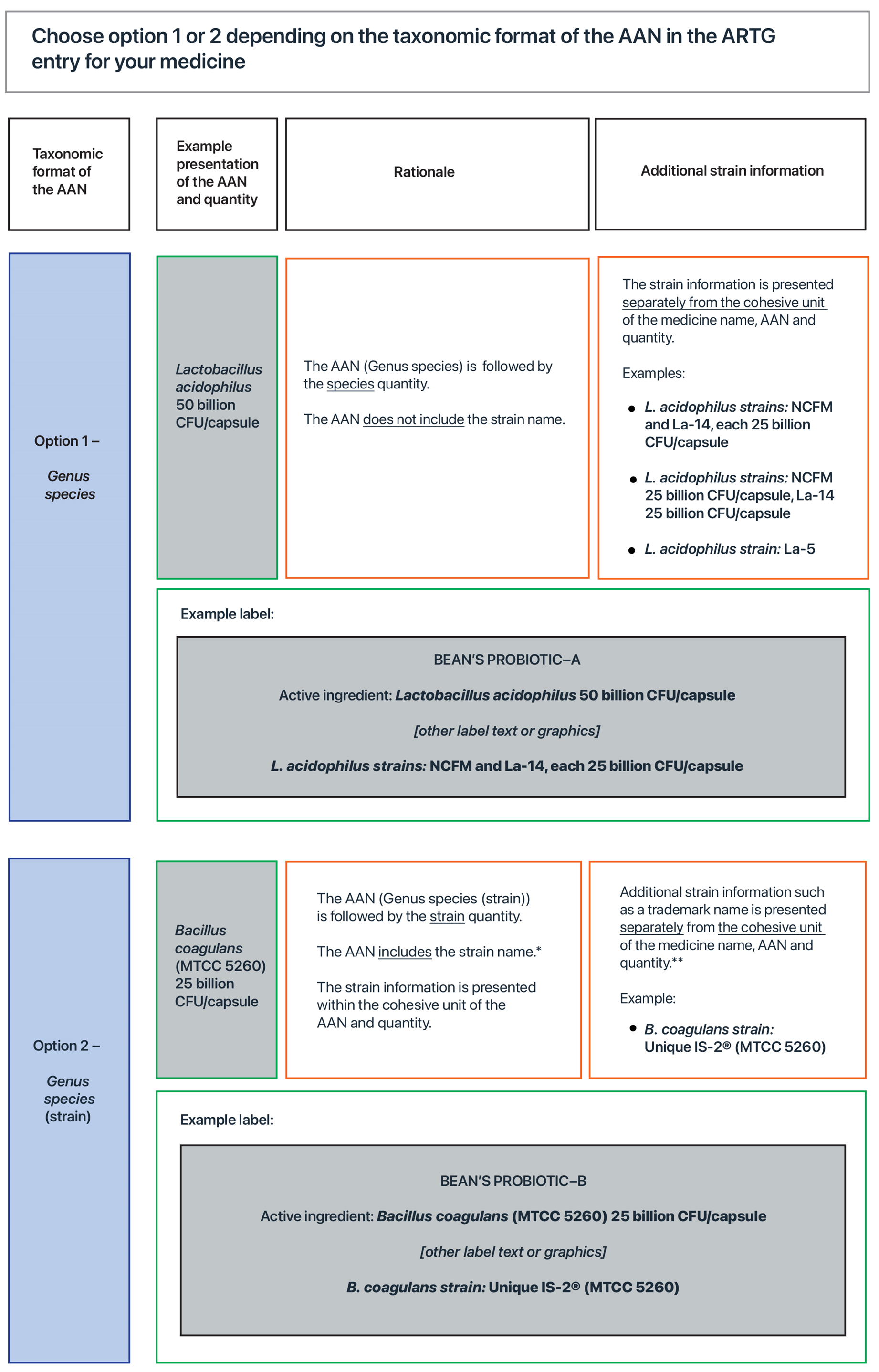
Compliance with both TGO 92 subsection 9(3) and the applicable default standard (either Ph. Eur./BP 3053 or USP–NF 64) can be achieved by presenting the strain name on the main label in a manner that does not disrupt the cohesive unit. The cohesive unit is when the name of the medicine and the names and quantities of the active ingredient(s) on the main label are together and not separated by any text or graphics.
Labels do not need to state that the medicine conforms to the USP or NF.
Figure 7 provides two options for how to present the ingredient name and quantity on the label. Option 1 will apply in most cases, although sponsors must use option 2 if it is applicable.
- Option 1 should be chosen if the name of the active ingredient (the AAN) in the ARTG entry for their medicine is in the taxonomic format ‘Genus species’. At the time this Guideline was published, all AANs for probiotics were in this format and therefore option 1 will apply in most cases.
- Option 2 should only be chosen if the name of the active ingredient (the AAN) in the ARTG entry for their medicine is in the taxonomic format ‘Genus species (strain)’. At the time this Guideline was published, no AANs for probiotics were in this format and therefore option 2 is included in this Guideline to cover potential changes to the AAN.
A transition period has been provided to sponsors to allow time for product labels to be compliant with existing requirements in relation to TGO 92 subclause 9(3) and either Ph. Eur./BP 3053 or USP–NF.
Labels of listed probiotic medicines that are covered by the consent under s14 of the Act are able to choose to comply with either TGO 92, Ph. Eur./BP 3053 or USP-NF until 1 October 2026. During this period, the active ingredients can be labelled at the species OR strain level.
After this period, labels are required to comply with the labelling ministerial standard AND the applicable default standard.
Units on the label for ingredient quantity
The expression of the quantity unit for an ingredient on the label must comply with the ministerial standard Therapeutic Goods Order No. 92 - Standard for labels of non-prescription medicines (TGO 92) which specifies in subparagraph 11(2)(i)(v) that for preparations containing biological organisms, the quantity or proportion of active ingredients is to be expressed as the number of organisms present per metric unit for liquids and powders and as the number of organisms present per dosage unit for other dosage forms.
This means that the number of organisms must be expressed on the label as a number per metric unit or per dosage unit in order to comply with TGO 92.
The unit of measure for quantifying an ingredient in a probiotic is entered into the Electronic Listing Facility (ELF) when applying to list a medicine on the ARTG. This unit is selected from a drop-down list of available units from the TGA Code Tables and reflects the unit expressed on the product specification, C of A and product label.
The TGA Code tables include thousand, million or billion of the following units (among others): organisms, organisms/g, organisms/mg, organisms/g, organisms/mL, CFU, CFU/g, CFU/mL.
A sponsor should carefully select the appropriate unit from those available for selection and not select units unsuitable for probiotic ingredients, such as 'mg/g' (even if they are included in a drop-down menu). For liquid probiotics, the unit ‘CFU per mL’ or ‘organism per mL’ is an option, while the metric unit ‘CFU per g ’or ‘organism per g’ would be more suitable for probiotics that are freeze dried.
If a non-culture-based method is used to enumerate the number of live/viable organisms (e.g. flow cytometry, qPCR with a nucleic acid stain or digital droplet PCR with strain specific PCR primers), then the enumeration must be reported in appropriate units that also comply with TGO 92. An example unit for a non-culture-based method is ‘billion organisms/g’[7]. The enumeration test method (stated in the product specification) will define what is tested and any calculations required to obtain the reported result for the number of organisms per metric unit or per dosage form. Although the unit 'active fluorescence units' (AFUs) is not an option, AFU represents, and can be converted to, the number of organisms (on the basis of ISO 19344|IDF 232 (2015) or as validated).
Any unit of proportion without a metric quantity that is available for selection from TGA Code tables—when viewed together with other information in the ARTG entry—is considered to be ‘per dosage unit’ as the dosage unit is specified elsewhere in the ARTG entry; e.g. ‘billion organisms per capsule’. The unit ‘billion organisms’, for example, is restricted to divided preparations, therefore the quantity 'billion organisms' in an ARTG entry is linked with a specific dosage unit, such as a capsule that the sponsor has selected in their application.
The successful validation of an application in ELF does not imply that a medicine is compliant with the legislative requirements. The listed medicines validation system is a tool to assist sponsors in listing a medicine on the ARTG. Sponsors are expected to be aware of, and comply with, all relevant regulatory requirements for their listed medicine.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Applicable legislation
This section details the legislative framework and requirements that a sponsor must comply with to control the quality of their listed probiotic medicine. This section provides the legislative basis for the information in section Demonstrating compliance with legislative requirements. The relationship between this section and section 4 is explained in the opening paragraphs of section 4.
This section is intended to assist with sponsor and manufacturer compliance by naming and explaining the most relevant legislation for ensuring the quality of probiotic medicines.
The Act
The Act (Therapeutic Goods Act 1989) provides a system of controls for the quality, safety and efficacy of therapeutic goods. To ensure the quality, safety and efficacy of their medicine, a sponsor must comply with the Act, and delegated (secondary) legislation under the Act, such as Regulations, Standards, Orders, Codes and Determinations.
There are legal consequences for sponsors who fail to control the quality, safety and efficacy of their medicine in accordance with legislative requirements. This may include cancellation and/or recall of the medicine, issue of infringement notices or formal court action. If a medicine is cancelled from the ARTG, it is an offence under the Act to manufacture, supply for use, import or export the medicine (subject to limited exceptions).
Non-compliance with Chapter 3 of the Act (including Part 3-1 ‘Standards’, Part 3-2 ‘Registration and listing of therapeutic goods’, and Part 3-3 ‘Manufacturing of therapeutic goods’) may incur civil penalties and criminal offences, unless stated otherwise (for example, where there is consent under sections 14/14A, or exemptions under sections 18 or section 34).
Refer to Table 3 for a list of the more relevant legislation for regulating quality, and for links to delegated legislation, pharmacopoeias, and TGA-adopted guidance.
Note that legislation other than that mentioned in these Guidelines may also be relevant to a sponsor’s particular situation. For general guidance on other legislation, refer to the TGA guideline Understanding listed and registered complementary medicine regulation.
| Section in these Guidelines | Therapeutic Goods Act 1989 (section or subsection) | Delegated legislation, relevant pharmacopoeia or guidance | |
|---|---|---|---|
| Listing certification | Listing of certain medicines – matters that applicant must certify | 26A(2) | |
| Conditions of listing | Conditions of listing – must be complied with. Non-compliance may lead to cancellation | 28 | See, also, Therapeutic Goods (Listed Medicines—Conditions of Listing) Determination 2022 |
| Cancellation of listing | Cancellation of listing – various grounds | 30(1), 30(1A), 30(2) | |
| Permissible Ingredients Determination | Permissible ingredients – listed medicines must only contain permissible ingredients | 26BB | Therapeutic Goods (Permissible Ingredients) Determination |
| Listing certification, Conditions of listing | Permissible indications – medicines listed (under s 26A) can only use permissible indications | 26BF | Therapeutic Goods (Permissible Indications) Determination |
| Standards | Definition of ‘default standard’ See, also, paragraphs (b), (c) and (d) of definition of ‘standard' | 3(1) | European Pharmacopeia (Ph. Eur.) |
| Special provisions relating to ministerial standards and default standards – particularly where there are inconsistencies or questions of application | 13 | ||
| Criminal offences, or civil penalty provisions, for importing, supplying or exporting goods that do not comply with standards | 14, 14A | ||
| Ministerial standards | Ministerial standards, orders | 10 See also 13, 14, 14A | Therapeutic Goods Order No. 92 - Standard for Labels of Non-prescription Medicines (TGO 92) Therapeutic Goods (Standard for Tablets, Capsules and Pills) (TGO 101) Order 2019 (TGO 101) |
| Good manufacturing practice | Manufacturing principles – to be observed in manufacture of therapeutic goods | 36 | Guide to Good Manufacturing Practice for Medicinal Products (PIC/S Guide to GMP) |
Listing certification
When an application is made to list a medicine under section 26A, the applicant must certify all matters under subsection 26A(2) of the Act, including that the information included in or with the application is correct (26A(2)(k) of the Act). If required by the Secretary (under subsection 31(2) of the Act), the applicant must be able to provide information or documents relating to their certifications. As such, the applicant must be able to demonstrate that their certifications are true and accurate. Where certifications are incorrect this may lead to cancellation.
The certifications most relevant to controlling the quality, safety and efficacy of probiotic medicines are included in Table 4.
| Paragraph of the Act | Certification |
|---|---|
| 26A(2)(b) | The medicine is safe for the purposes for which it is to be used. |
| 26A(2)(ca) and (cb) | The medicine does not contain an ingredient that is not specified in a determination under paragraph 26BB(1)(a); and if a determination under paragraph 26BB(1)(b) specifies requirements in relation to ingredients being contained in the medicine—none of the requirements have been contravened. Note: This certification relates to compliance with the Permissible Ingredients Determination. |
| 26A(2)(d) | The medicine conforms to every standard (if any) applicable to the medicine. Note: Standards include ministerial standards such as TGO 92 and TGO 101, and the default standards Ph. Eur., BP and USP–NF. |
| 26A(2)(f) | The medicine complies with all prescribed quality or safety criteria applicable to the medicine.* |
| 26A(2)(fc) | The applicant holds information or evidence showing the medicine’s specifications will be maintained under the conditions set out on the medicine’s label until the medicine’s expiry date |
| 26A(2)(fd) and (fe) | Each indication proposed to be accepted in relation to the inclusion of the medicine in the Register is covered by a determination under paragraph 26BF(1)(a); and if a determination under paragraph 26BF(1)(b) specifies requirements in relation to an indication proposed to be accepted in relation to the inclusion of the medicine in the Register—none of the requirements have been contravened. Note: This certification relates to compliance with the Permissible Indications Determination. |
| 26A(2)(ja) | The applicant holds information or evidence to support each indication proposed to be accepted in relation to the inclusion of the medicine in the Register; and the information or evidence complies with any requirements specified in a determination under subsection (2B). |
| 26A(2)(e), (h) and (i) | If the medicine has been manufactured in Australia—each step in the manufacture of the medicine has been carried out by a person who is the holder of a licence to carry out that step; and all the manufacturers of the medicine are nominated as manufacturers in the application; and the applicant has, with manufacturers of the medicine who are manufacturers of the prescribed kind, written agreements containing such matters as are prescribed.* |
| 26A(2)(k) | The information included in or with the application is correct. |
| * At the time these Guidelines were prepared there was nothing prescribed in the Therapeutic Goods Regulations 1990 for the purposes of paragraphs 26A(2)(f) or 26A(2)(i). | |
Conditions of listing
Some conditions apply automatically to listed medicines and are imposed by section 28 of the Act (through the Therapeutic Goods (Listed Medicines—Conditions of Listing) Determination 2022). Conditions of listing are also able to be imposed, as a matter of discretion, by a delegate under section 28.
Under subsection 28(6) of the Act, a sponsor must (at any time while their medicine remains listed) provide information or evidence to the Secretary, if asked to do so, that supports a claim in relation to their medicine (other than a claim that is an indication). This can include claims about the shelf life of the medicine. The information or evidence must be held at the time of listing and retained at all times while the medicine remains listed.
Another condition of listing is that under subsection 28(7) of the Act. The sponsor must (at any time while the medicine remains listed) provide information or evidence to the Secretary, if asked to do so, that supports an indication that is accepted in relation to the inclusion of the medicine in the Register. This information or evidence must be held at all times while the medicine remains listed. While this condition is typically associated with literature evidence (such as clinical trial articles), the information or evidence may also include quality-related data that demonstrates the manufactured product can provide the necessary active ingredients at the correct dose in alignment with the literature evidence used to support the efficacy of the medicine.
Cancellation of listing
Listed probiotic medicines that are found to be non-compliant under provisions controlling quality may be cancelled from the ARTG and can no longer be sold in Australia. Examples of cancellation provisions relevant to controlling quality are included in Table 5.
| Paragraph of the Act | Cancellation provision |
|---|---|
| 30(2)(a) | The Secretary may cancel the listing of the goods from the ARTG if it appears to them that the quality, safety or efficacy of the goods is unacceptable. |
| 30(2)(ba) | The Secretary may cancel the listing of the goods from the ARTG if it appears to the Secretary that any of the certifications under paragraphs 26A(2)(b), (c), (d), (da), (f), (fa), (fb), (fc), (h), (i), (j), (ja) or (k) or subsection 26A(2A) are incorrect. |
| 30(2)(c) | The Secretary may cancel the listing of the goods from the ARTG if the sponsor has refused or failed to comply with a condition to which the inclusion of the goods is subject (other than the condition under paragraph 28(5)(d)). Note: Refer to section Conditions of listing for information about more relevant conditions of listing. |
Permissible Ingredients Determination
Listed medicines must only contain ingredients that are in the Therapeutic Goods (Permissible Ingredients) Determination (the Permissible Ingredients Determination) and they must not contravene any of the requirements for an ingredient.
When applying to list a probiotic medicine on the ARTG, each active microbial ingredient permitted for use in the Permissible Ingredients Determination and available for selection on the ARTG must be at the appropriate taxonomic level of strain or species relied upon for the claimed efficacy. Refer to Figure 3 for options when the efficacy evidence is held for a strain, but only species are available for selection when listing a medicine on the ARTG.
Active microbial ingredients available for selection are specified in Column 2 of the Permissible Ingredients Determination. Column 4 may also include specific requirements permitting the use of certain strains for that species, however these strains are not available for selection as separate ingredients when listing a medicine on the ARTG (refer to section 5 in the Determination).
If a strain (or species) is not in the Permissible Ingredients Determination, then an application for evaluation of a new substance can be submitted to the TGA. See Requirements for microorganism characterisation for Listed Medicines and Registered Complementary Medicines and Application requirements for new substances in listed medicines.
If an active microbial ingredient is permitted for use in the Permissible Ingredients Determination, there will be a corresponding Australian Biological Name (ABN) in the Ingredients Table in the TGA Business Services (TBS) portal for selection during a listing application. For more information about ABNs, see Ingredients in therapeutic goods and Naming convention for biological substances.
Standards
Therapeutic goods supplied in, exported from or imported into Australia must comply with applicable standards (Part 3-1 of the Act). Criminal offences and civil penalties apply to persons who do not comply and who otherwise do not have consent (sections 14/14A of the Act). For more information refer to the guidance Compliance with ministerial and default standards.
When an application is made to list a medicine under 26A(1) of the Act, the applicant must certify that the medicine conforms to every standard (if any) applicable to the medicine (26A(2)(d) of the Act). Certifications must be correct. As noted above, the Secretary may cancel the listing of the medicine from the ARTG if it appears to the Secretary that the certification is incorrect (30(2)(ba) of the Act).
There may be multiple default standards (section Default standards) and ministerial standards (section Ministerial standards) that can apply to a listed probiotic medicine. Refer to section How to determine which quality standards to comply with.
Default standards
A default standard in relation to therapeutic goods, means that if the goods are the subject of one or more monographs in the British Pharmacopoeia (BP), the European Pharmacopoeia (Ph. Eur.), or the United States Pharmacopeia–National Formulary (USP–NF), then the statements in the monograph(s) (except for statements or monographs exempted by the Minister), are interpreted in accordance with the General Notices section of the relevant pharmacopoeia.
The default standards are regularly updated, therefore sponsors should refer to the relevant text in the current pharmacopoeia in full.
Compliance with default standards
Section 13 of the Act relates to the application of ministerial standards and default standards in particular circumstances. Importantly, controlling the quality of active microbial ingredients must be in accordance with at least one default standard to which a medicine is subject (subsection 13(7) of the Act).
For a probiotic medicine, the most relevant monograph from the default standards is Ph. Eur./BP 3053[8], which is the abbreviated term used herein for the European and British Pharmacopoeias, general monograph 3053 ‘Live biotherapeutic products for human use’ (Official in April 2019).
Another relevant monograph from the default standards is USP–NF 64, which is the abbreviated term used herein for the United States Pharmacopeia National Formulary, general chapter 64 ‘Probiotic tests’ (Official on 1 August 2019). The USP–NF 64 applies to a probiotic medicine if there is a USP–NF individual monograph that is relevant and refers to USP–NF 64. For example, the individual USP–NF monograph for Lactobacillus acidophilus (Strains La-14 or NCFM) refers to USP–NF 64. Refer to section USP–NF for further details.
To determine which one of these default standards has applicable requirements, and therefore which quality standards apply to a final product, refer to section How to determine which quality standards to comply with.
This means that because:
- compliance must be in accordance with at least one default standard, and
- there are currently very few USP–NF individual monographs that are relevant to listed probiotic medicines,
generally Ph. Eur./BP 3053 must be complied with.
Each relevant default standard applies in its entirety. A medicine does not comply with an entire default standard if a requirement within cannot be performed or met. The general notices sections of Ph. Eur., BP and USP–NF express that all requirements in relevant individual monographs (and relevant applicable general monographs or general chapters) must be met to demonstrate compliance with the standard. Readers should refer to the relevant text in the current pharmacopoeia in full.
An individual monograph may apply to a medicine or an ingredient even when the title of the monograph is not identical to the name of the medicine or ingredient. Where the name of a medicine or ingredient is a variant of the name (e.g. a transposed/inverted name or synonym as referred to in the USP-NF and BP General Notices) of an individual monograph, the provisions of the monograph, including definitions, and the General Notices section of the relevant pharmacopoeia should be reviewed to determine whether the monograph applies. For example, consider whether the name of an ingredient in your probiotic medicine is a variant name of a strain in a USP–NF monograph and thus whether the ingredients are materially the same and whether that monograph is applicable to your medicine.
Ph. Eur. and BP
Ph. Eur. and BP standards contain general monographs and individual monographs. A therapeutic good (ingredients/substances or final products) may be the subject of general and/or individual monographs—even when cross references to applicable general monographs are not given in individual monographs.
Table 6 is a broad outline of requirements in the Ph. Eur./BP 3053 and the general notices sections in these pharmacopoeias. Readers should refer to the relevant parts of the current version of the pharmacopoeia in full.
| Pharmacopoeia Section heading | Identification | Quantification |
|---|---|---|
| Final lot |
|
|
| Labelling | The label must state the name of each strain. | The label must state the quantity of each strain (CFU/g, CFU/mL, CFU/unit or viable cells/mL). |
| Stability | The Ph. Eur. and BP ‘General notices’ section 1.1 requires a preparation to comply throughout its period of validity, meaning that the medicine must comply with the quantification requirements for the duration of the product’s shelf life. | |
| Characterisation (of starting material) |
| |
Alternative approach to compliance
A medicine is of Ph. Eur. quality and BP quality if it complies with all of the requirements stated in the monograph (Ph. Eur. ‘General notices’, section 1.1.2.2; and BP ‘General notices', Part II). In brief, these provisions also explain that when a manufacturer is assessing compliance with Ph. Eur./BP, they are not obliged to perform all of the tests in a monograph before release. The manufacturer may assure themselves that a product is of pharmacopoeial quality by other means, such as by design together with control strategy and data derived, for example, from validation studies of the manufacturing process.
Note that such alternative means of assurance, such as using a quantified by input approach (refer to section Quantified by input ), should be scientifically justified. Such as with data from process validation or a history of compliant testing that is complemented by no changes to process or formulation. Also note that for critical quality attributes, all necessary and relevant tests for quality control must be carried out before release (refer to section GMP – Testing for quality).
In using this alternative approach to compliance, the manufacturer would be assuring conformance with the intended outcome of the Ph. Eur./BP 3053 (for example, ensuring the identity and quantity of each strain in the final lot, and use of a suitable microbial enumeration test). Examples of ways in which probiotic sponsors/manufacturers can achieve this is further explained in section Demonstrating compliance with legislative requirements.
Ultimately the sponsor is responsible for complying with the requirements in the Ph. Eur./BP 3053 and being able to provide sufficient information or evidence (including details of manufacturing assurances) to demonstrate such compliance. Also note that alternative analytical methods other than those described in the Pharmacopoeia may be employed for routine and control purposes (refer to section Alternative tests for identification and quantification).
Microbial contamination
Ph. Eur./BP 3053 refers to tests and acceptance criteria for enumeration of microbial contaminants.[9] Tests include those referred to in Chapter 2.6.36 ‘Microbiological examination of live biotherapeutic products: Tests for enumeration of microbial contaminants’, and in Chapter 2.6.38 ‘Microbiological examination of live biotherapeutic products: Tests for specified microorganisms’. In addition, Chapter 5.1.6 ‘Alternative methods for control of microbiological quality’ may be used for qualitative, quantitative and identification tests of contaminant microorganisms.
USP–NF
The applicability of USP–NF to a probiotic medicine is subject to the following:
- Only official articles (official substances e.g. dietary ingredients, or official products e.g. dietary supplements) for which there is a monograph can comply with USP–NF.
- At the time these Guidelines were prepared, monographs exist for single strain substances (such as a starting material ingredient or a product that contains a single strain) but not for multi-strain substances or products. Additionally, although the USP monograph title is at the species level, the monograph only applies to the strains that are specifically referred to in the monograph. Nevertheless, refer to the current version of the pharmacopoeia for updated information.
- If there is an applicable individual monograph (such as for a starting material or a single strain product), then strain-level identification, quantification and labelling must be in accordance with the requirements in the individual monograph, applicable general chapters that are cited in that individual monograph and the ‘General notices and requirements’.
- Examples of quantification and labelling requirements in an individual monograph include:
- The acceptance criterion for quantification: no less than 100% of the labelled viable cell count, in CFU/g (or CFU/capsule), of the specified strain.
- The labelling requirement: the ingredient should be labelled with the genus, species and strain name(s) and with the formulated enumeration in CFU/g, CFU/dose, dose/g or CFU/capsule.
- USP–NF 64 is only applicable to a microbial ingredient when USP–NF 64 is cited in the individual monograph for the ingredient or product.
- For USP–NF 64 to become applicable to a multi-strain product, an individual monograph would first need to be developed and it would need to cite USP–NF 64.
- If an individual monograph procedure or acceptance criteria is different to that in
USP–NF 64, then the product is exempt from complying with that portion of USP–NF 64.
An example of how the requirements in an individual monograph may cite the requirements in USP–NF 64 is in Table 7. Table 8 provides a summary of the requirements in USP-NF 64. Readers should refer to the relevant parts of the current version of the pharmacopeia in full.
Section in an individual monograph
| Example requirements (Always check the current relevant individual monograph) |
|---|---|
| Identification | Use either microscopic identification or nucleic acid-based identification to strain level. |
| Acceptance criteria | Presents as rods. Amplification product of 184 base pairs. There should be no amplification product of 600 base pairs. |
| Enumeration | See Probiotic Tests <64> Enumeration, Enumeration for non-spore-forming bacteria strains. Note: some individual monographs provide a procedure and do not refer to <64>. |
| Acceptance criteria | NLT 100% of the labelled viable cell count, in CFU/g of the strain. |
| Contaminants |
|
| Labelling | Ingredient should be labelled with the genus, species and strain names and with the formulated enumeration in CFU/g. |
| Pharmacopoeia Section heading | Identification | Quantification |
|---|---|---|
| Final lot |
|
|
| Labelling |
|
|
Microbial contamination
USP–NF 64 recommends tests and acceptance criteria for contaminant microorganisms, including <2021> ‘Microbial enumeration tests’ and <2022> ‘Microbiological procedures for absence of specified microorganisms—nutritional and dietary supplements’.8 In addition, guidance on selection, evaluation and use of microbiological methods as alternatives to compendial methods is provided in <1223> ‘Validation of alternative microbiological methods’.
Alternative tests for identification and quantification
An alternative analytical test or procedure to those presented in the relevant default standards must be appropriately validated to demonstrate that it is suitable for its intended purpose. The TGA guideline Finished product (medicine) analytical procedure validations for complementary medicines provides further information.
Ph. Eur./BP 3053 requires suitable methods to be used for identification and quantification at the level of strain. In brief, the ‘General notices’ of the Ph. Eur., at section 1.1.1.2, defines ‘suitable’ (if criteria for suitability are not described in the text) to be when suitability is demonstrated to the satisfaction of the competent authority (e.g. the TGA). An example would be any appropriately validated strain-level test.
Ph. Eur. ‘General notices’, at section 1.1.2.5, also outlines that alternative methods may be used for control purposes—providing the methods enable an unequivocal decision regarding whether compliance with the standards of the monographs would be achieved if the official methods were used.
BP ‘General notices’, Part II (on ‘Assays and tests’), also allows for analytical methods other than those described in the Pharmacopoeia to be employed for routine purposes.
BP ‘Supplementary Chapter 1 H’ (on ‘Biological assays and tests’) explains that the methods of biological assay described in the Pharmacopoeia have been found satisfactory but will not necessarily be the best methods for use in all circumstances. In most instances, they may be replaced by other methods if it can be shown that such methods are at least equally accurate and precise and provide a measurement of the same active principles.
USP–NF allows alternative methods or procedures that have been fully validated and produce comparable results to the compendial method or procedure within allowable limits. Refer to USP–NF <1225> ‘Validation of compendial procedures’.
USP–NF 64 does not provide alternative methods for identification. However, for quantification of contaminant microorganisms, USP–NF 64 provides for alternative microbiological procedures (including automated methods) if their equivalence to the pharmacopoeial procedure has been demonstrated. Refer to USP–NF <1223> ‘Validation of alternative microbiological methods’.
Relationship with ministerial standards
If there is an inconsistency between an applicable default standard and an applicable ministerial standard (such as the Therapeutic Goods Orders referred to in sections TGO 92 – Labelling and TGO 101 – Dosage forms that are tablets, capsules and pills),then the inconsistent requirement in the default standard is to be disregarded, not the entire default standard (subsection 13(2) of the Act).
Strain names and quantities are to be stated on the label in accordance with Ph. Eur./BP 3053 and USP–NF 64. This must be done in a manner that does not contravene the requirements in TGO 92. Refer to sections Labelling and TGO 92 – Labelling.
Ministerial standards
A ministerial standard is also known as a Therapeutic Goods Order (TGO).
Therapeutic Goods (Microbiological Standards for Medicines) (TGO 100) Order 2018, section 11 ‘Microbiological attributes of a non-sterile medicine’, is not applicable to probiotic medicines. Section 11 references test methods within Ph. Eur., BP and USP–NF that explicitly state they are not applicable to products that contain viable microorganisms as active ingredients (refer to Ph. Eur./BP 2.6.12 (which is a procedure in Ph. Eur./BP 2.6.13) and USP–NF 61 (which is a procedure in USP–NF 62). This means, those methods in section 11 of TGO 100 are not suitable for detecting microbial contamination (bioburden) in the presence of active probiotic ingredients.
Instead, for probiotics, refer to the tests and acceptance criteria (published more recently, in 2019) for enumeration of microbial contaminants in either Ph. Eur. or USP–NF (sections titled Microbial contamination).
TGO 92 – Labelling
The Therapeutic Goods Order No. 92 - Standard for Labels of Non-prescription Medicines (TGO 92) requires, amongst other details, that the names (and quantities or proportions) of all active ingredients in a medicine be included on the medicine’s label (see section 8, especially paragraph 8(1)(b) and (c) and sections 9 and 10 of TGO 92).
The ‘name of an active ingredient’ is defined, as relevant, in section 6 of TGO 92 to mean the name that is accepted for inclusion in the AAN list that can be searched in the Ingredients Table in the TGA Business Services website. These ingredients are named according to international scientific naming conventions to ensure their identities are unique, unambiguous and thus distinguishable.
For further guidance see Medicine labels: Guidance on TGO 91 and 92.
TGO 101 – Dosage forms that are tablets, capsules and pills
The Therapeutic Goods (Standard for Tablets, Capsules and Pills) (TGO 101) Order 2019 applies to listed (and registered) medicines in dosage forms that are tablets, capsules or pills. The section below focusses on the requirements for tablets and capsules, not pills.
Consider whether other ministerial or default standards are applicable to different dosage forms (e.g. powders, oil drops [pearls], chewable tablets and gummies).
Applicable monographs
The requirements for tablets and capsules outlined in section 8 of TGO 101 depend on whether or not there is an applicable monograph.
In brief, an applicable monograph is defined in subsection 4(1) of TGO 101 to mean a default standard for a preparation or product in the Ph. Eur., BP or USP–NF that comprises:
- a specific monograph (called an individual monograph in this guideline and in the Ph. Eur., BP and USP–NF),
- one or more applicable general monographs, and
- one or more applicable general chapters,
interpreted in accordance with the general notices of the relevant pharmacopoeia.
An applicable monograph exists if there is an individual monograph in the Ph. Eur, BP or USP–NF to which the probiotic tablet or capsule is subject. An applicable monograph is comprised of an individual monograph together with any applicable general monographs or general chapters. The general notices of a pharmacopoeia assist with determining when a general monograph or general chapter is applicable.
Ph. Eur./BP 3053 and USP–NF 64 are not applicable monographs for the purposes of TGO 101 by themselves because they are not individual monographs, instead they are a general monograph and a general chapter, respectively. Although, as explained in other sections, the requirements in Ph. Eur./BP 3053 will still be applicable to probiotic tablets and capsules; and the requirements in USP–NF 64 will be applicable where a probiotic tablet or capsule is subject to an individual USP–NF monograph that refers to USP–NF 64.
For further guidance see Guidance for TGO 101.
Ph. Eur. or BP individual monographs
The Ph. Eur. and BP do not include any individual monographs for probiotic ingredients (at the time these Guidelines were prepared); therefore, there are no applicable monographs in Ph. Eur. or BP for the purposes of TGO 101.
USP–NF monographs for dietary ingredients or dietary supplements
Monographs for dietary ingredients or dietary supplements in the USP–NF are considered to be individual monographs, and therefore can be applicable monographs relevant to probiotic tablets and capsules for the purposes of TGO 101 (together with any applicable general monographs or general chapters).
The USP–NF includes individual monographs for probiotic species and specific strains. For example, the individual USP–NF monograph for Lactobacillus acidophilus (for strains La-14 or NCFM) would be an applicable monograph where relevant.
However, individual monographs for a probiotic species and strain are only applicable to formulated products (or starting materials) that contain a single strain—unless explained otherwise in the ‘Definition’ section of the monograph. Apart from this exception, an applicable monograph for a multi-strain probiotic would require an individual monograph for the formulated product.
A USP–NF general chapter can also be relevant to a tablet or capsule if there is a relevant USP–NF individual monograph that refers to the general chapter (such as the reference to the general chapter USP–NF 64 in the individual monograph for Lactobacillus acidophilus). With the exception that a general chapter is not relevant if an individual monograph refers to it for information purposes only (Section 3.10 of the USP–NF General notices).
Requirements for tablets and capsules
Figure 2 provides a decision tool that summarises the following and assists sponsors with determining which parts of TGO 101 and default standards to comply with.
If an applicable monograph exists (there can be more than one), subsection 8(1) of TGO 101 specifies that a sponsor can elect to comply with that applicable monograph, subject to the matters specified in Division 2 (of Part 2 of TGO 101). Alternatively, a sponsor can elect to comply with the Australian specific requirements in Division 3 (of Part 2 of TGO 101) and the requirements relevant to the tablet or capsule specified in one of the general monographs/chapters of the Ph. Eur., BP or USP–NF.
In the absence of an applicable monograph, subsection 8(2) of TGO 101 specifies that a medicine must comply with the Australian specific requirements in Division 3 (of Part 2 of TGO 101) and the requirements relevant to the tablet or capsule specified in one of the general monographs/chapters of the Ph. Eur., BP or USP–NF.
In either case, when Division 3 applies, it applies together with the requirements specified in one of the general monographs or general chapters that is relevant to the tablet or capsule (from either the Ph. Eur., BP or USP–NF).
Not less than the stated content
The Australian specific requirements in Division 3 (of Part 2 of TGO 101) provide, among other things, that the assay limit for an active probiotic ingredient in a probiotic tablet or capsule must be ‘not less than the stated content’ (subsection 14(2) and Schedule 2). The stated content means the quantity of the active ingredient that is stated on the label and is thus claimed to be present in each capsule (or tablet) (subsection 4(1) of TGO 101).
Good manufacturing practice
When an application is made to list a medicine under section 26A, the applicant must certify matters in relation to manufacturing under paragraphs 26A(2)(e), (h) and (i) of the Act (refer to section Listing certification in these Guidelines).
Manufactured therapeutic goods, and the person manufacturing the therapeutic goods, must comply with Part 3-3 of the Act ‘Manufacturing of therapeutic goods’. Good Manufacturing Practice (GMP) is a system that ensures the quality of a medicine is consistently and adequately controlled. The PIC/S Guide to GMP for Medicinal Products is legally enforced in Australia through section 36 of the Act and the Therapeutic Goods (Manufacturing Principles) Determination 2020 (the Manufacturing Determination).
The PIC/S Guide to GMP includes controls for the quality of probiotic medicines such as ongoing stability (e.g. Chapter 6), starting materials (e.g. Chapter 1), supplier qualification (e.g. Chapter 5), product Quality Reviews (e.g. Chapter 1), and manufacturing validations and equipment qualification requirements (e.g. Annex 15).
Where the PIC/S Guide to GMP states that a procedure or requirement ‘should’ be followed, the manufacture of therapeutic goods must follow the procedure or requirement in order to comply, subject to certain exceptions (Manufacturing Determination, Schedule 1, Part 1(2)).
The exceptions allow for a particular procedure or requirement not to be adopted, or an alternative to be adopted. However, the manufacturer must demonstrate that this will not increase the risk that the goods could (or could potentially) cause harm or injury, will not increase the risk of the goods failing to comply with an applicable standard or relevant condition of listing, and will not depart from any applicable record keeping requirements contained in the PIC/S Guide to GMP.
Voluntary procedures or requirements specified in an Annex to the PIC/S Guide to GMP do not need to be complied with (for example, Annex 20).
On-going stability
On-going stability testing of listed probiotic medicines must comply with the applicable procedures and requirements in Chapter 6 ‘Quality Control’ of the PIC/S Guide to GMP for Medicinal Products - Part I. See the TGA guideline On-going stability testing for listed and complementary medicines for technical guidance.
An on-going stability program monitors a medicine over its shelf life to determine that it remains, and can be expected to remain, within its specifications under the labelled storage conditions (PIC/S Guide to GMP, Part 1, Chapter 6). The stability program for a medicine must be described in a written protocol, which must include (among other things) relevant test methods and acceptance criteria. This ensures that a probiotic medicine will deliver the required quantity of each active ingredient strain (or species if applicable) per dose throughout the shelf life (in accordance with a sponsor’s certification under paragraph 26A(2)(fc) of the Act).
Testing for quality
Quality Control ensures that tests are carried out for all critical quality attributes, and that materials are not released for use, sale or supply until their quality is satisfactory. This requires validated test methods (PIC/S Guide to GMP, Part 1, Chapter 1), using approved testing methods for the product (Chapter 6), recording data with references to specifications and testing procedures (Chapters 4 and 6), and test method transfer (e.g. between laboratories) requirements (Chapter 6).
For guidance on test method validation see Finished product (medicine) analytical procedure validation for complementary medicines, Starting material analytical procedure validation for complementary medicines and in the ICH quality guidelines.
Starting materials
The starting materials for a probiotic medicine include all active microbial ingredient strains (and excipients) that are input at the start of manufacturing. Starting materials are regulated and quality controlled through a system of vendor qualifications, supply chain traceability, approved specifications and materials testing. The selection, approval, qualification and maintenance of suppliers of starting materials, together with materials purchase and acceptance, must be documented as part of the pharmaceutical quality system (PIC/S Guide to GMP, Part 1, Chapter 5).
Manufacturers of finished products are responsible for testing starting materials in accordance with the product specifications (Chapter 5). They can utilise partial or full test results from the approved starting material manufacturer but must at a minimum include identification testing of each batch of starting material in accordance with Annex 8 of PIC/S Guide to GMP. However, identification testing alone would not be sufficient unless supported by comprehensive supplier approval processes (as outlined in Annex 8) and verification of all other quality attributes in the certificate of analysis (C of A). Identification of starting materials for active microbial ingredients provides assurance that the correct probiotic strain is used during manufacturing.
The TGA’s expectations in relation to the requirements for starting materials in the PIC/S Guide to GMP are explained in section Quality of starting materials.
Glossary
| AAN | Australian Approved Name |
| ABN | Approved Biological Name |
| ARTG | Australian register of therapeutic goods |
| BP | British Pharmacopoeia |
| C of A | Certificate of analysis |
| CFU | Colony forming units |
| Ph. Eur. | European Pharmacopoeia |
| GMP | Good Manufacturing Practice |
| ICH | International council for harmonisation of technical requirements for pharmaceuticals for human use |
| PIC/S | Pharmaceutical inspection convention and Pharmaceutical inspection co-operation scheme |
| Postbiotics | Medicines with therapeutic activity attributed to distinct ingredients that are inactivated, non-viable microorganisms and/or their components. Also known as paraprobiotics |
| Probiotic | live microorganisms that when administered in adequate amounts, are proposed to confer a health benefit on the host |
| QBI | Quantified by input |
| Species-level taxonomy | Refers to naming the Genus and species together, including subspecies (ssp) when relevant. |
| Strain-level taxonomy | refers to naming the Genus, species and strain together. |
| the Manufacturing Determination | Therapeutic Goods (Manufacturing Principles) Determination 2020 |
| the Permissible Ingredients Determination | Therapeutic Goods (Permissible Ingredients) Determination |
| TGO 101 | Therapeutic Goods (Standard for Tablets, Capsules and Pills) (TGO 101) Order 2019 |
| the Act | Therapeutic Goods Act 1989 |
| TGO 92 | Therapeutic Goods Order No. 92 - Standard for Labels of Non-prescription Medicines |
| TGO 100 | Therapeutic Goods (Microbiological Standards for Medicines) (TGO 100) Order 2018 |
| the Regulations | Therapeutic Goods Regulations 1990 |
| USP–NF | United States Pharmacopeia–National Formulary |
Footnotes
Medicines and foods are regulated differently. The Food–Medicine Interface Guidance Tool can help to determine whether a probiotic product is a food or a medicine.
General notices and requirements (official as of 1 December 2022), section 5.4 Identification.
'Beans probiotic' is a fictional product used for demonstration purposes.
At the time these Guidelines were prepared there were no individual monographs in the USP–NF for multi-strain products and there was only one individual monograph for a single strain product: ‘Bacillus coagulans capsules’. Therefore, nearly all individual monographs are for single strain substances (such as a starting material ingredient or a product that contains a single strain). However, readers should refer to the relevant text in the current pharmacopoeia in full.
A compendial method or procedure is a method or procedure that is described in the USP–NF.
All the information or evidence a sponsor has relied upon to determine their medicine’s shelf life and demonstrates that the medicine’s specifications will be maintained until expiry.
The default standards currently have no specific option for labelling with viable cells (or ‘organisms’) 'per dose', or 'per g' for example, they only state ‘per mL’. If such units (e.g., organisms/dose) are compliant with TGO 92 and are appropriate to the probiotic, then they may be expressed on the label. This is because subsection 13(2) of the Act provides that a requirement applicable to the goods in the default standard is to be disregarded in so far as it is inconsistent with the ministerial standard. For example, only the inconsistent requirement in the applicable default standard, viable cells/mL, is to be disregarded and organisms/dose may be used.
The general monographs Ph. Eur. 3053 and BP 3053 are harmonised and are thus expressed herein as Ph. Eur./BP 3053. Texts from the Ph. Eur., BP and USP–NF pharmacopoeias may be ‘harmonised’ (developed into a global quality standard) with another pharmacopoeia, meaning they have the same technical requirements (unless stated otherwise in their text).
Note also, Therapeutic Goods Order No. 100 (Microbiological Standards for Medicines) does not apply to probiotic medicines as the Ph. Eur./BP and USP–NF test methods referred to in TGO 100 are not applicable to medicines that contain viable microorganisms as active ingredients. Refer to section Ministerial standards.
Page history
Table 6 edited to remove the words "the label must state the name of any stabilisers and other excipients" in the labelling section.
Original publication
Table 6 edited to remove the words "the label must state the name of any stabilisers and other excipients" in the labelling section.
Original publication