Understanding requirements for unapproved therapeutic vaping devices and accessories in Australia
Guidance on the Therapeutic Goods (Medical Device Standard —Therapeutic Vaping Devices) Amendment Order 2024.
Purpose
The purpose of this guidance is to help sponsors and manufacturers of ‘unapproved’ therapeutic vapes understand the minimum quality and safety requirements for these devices.
This guidance is limited to information on the requirements that apply to ‘unapproved’ therapeutic vapes under MDSO 2024 and is only applicable to therapeutic vapes that are indicated for smoking cessation or the management of nicotine dependence.
Note
The requirements set out in the MDSO 2024 are not expected to be permanent. The vaping reforms are staged and the regulatory requirements for vapes are expected to continue to increase in the future.
It is anticipated that all therapeutic vaping devices will be required to meet Class IIb medical device requirements in the future and be included in the Australian Register of Therapeutic Goods (ARTG).
To include the device in the ARTG sponsors will need to have evidence that the device complies with the Essential Principles (EPs) for medical devices and that they hold relevant Conformity Assessment evidence.
Legislation
Note
This information is provided for guidance only.It should not be relied on to address every aspect of the relevant legislation (state, territory and federal). You should seek your own independent legal advice to ensure that all legal requirements are met.
For assistance with regulatory requirements, you may wish to engage a regulatory affairs consultant who is familiar with requirements for vaping goods.
What this guidance does not cover
This guidance does not include information on the requirements that apply to ‘unapproved’ therapeutic vaping substances, therapeutic vaping substance accessories, therapeutic vaping packs, and therapeutic vaping kits.
These products are subject to the provisions of the Therapeutic Goods (Standard for Therapeutic Vaping Goods) (TGO 110) Order 2021 (TGO 110).
For more information about how the TGO 110 applies to these vaping goods, refer to Guidance on the TGO 110.
This guidance does not contain information about therapeutic cannabis vapes including cannabis vaping devices and cannabis vaping device accessories.
For more information about therapeutic cannabis vapes, refer to the website on Cannabis vapes: Information for importers.
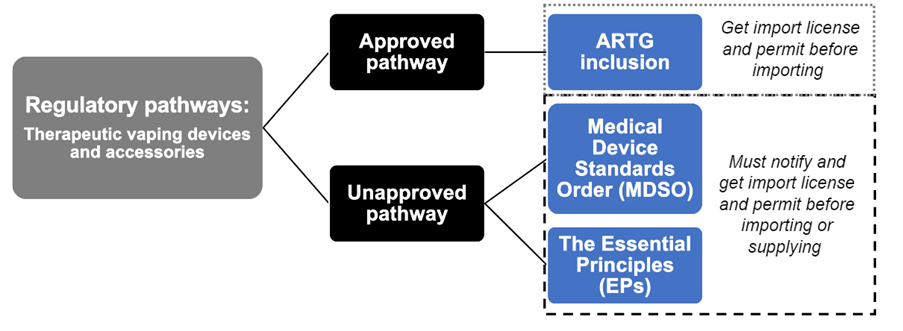
Regulatory pathways
There are two pathways for the supply of therapeutic vaping devices and accessories intended for smoking cessation or the management of nicotine dependence (See Figure 1).
Figure 1: Regulatory pathways for therapeutic vaping devices and accessories.
{kind=link}
Regulatory pathways: Therapeutic vaping devices and accessories - Approved pathway - ARTG inclusion - get import license and permit before importing.
Regulatory pathways: Therapeutic vaping devices and accessories - Unapproved pathway - Medical Device Standards Order (MDSO) - The Essential Principles (EPs) - Must notify and get import license and permit before importing or supplying.
These are:
- Approved pathway: the therapeutic vaping device and accessories are included in the Australian Register of Therapeutic Goods (ARTG), or
- Unapproved pathway: the therapeutic vaping devices and accessories are not included in the ARTG and must comply with either:
- the MDSO 2023, or
- the Essential Principles (EPs) which are the safety and performance requirements for medical devices.
You can only use the MDSO for your therapeutic vaping devices and accessories if they were not exclusively for the administration of a medicine before the commencement of MDSO 2023, on 1 March 2023.
If the MDSO does not apply to your products you must be able to demonstrate that they comply with the EPs, along with the conditions of the exemption (item 2.17 of Schedule 4 of the Therapeutic Goods (Medical Devices) Regulations 2002). You can find the conditions of the exemption in Appendix A of this guidance. For more guidance on how to comply with EPs, refer to Standards for therapeutic vaping devices checklist.
Detailed information about the regulatory requirements and pathways for different therapeutic vaping devices and accessories, including about the MDSO 2023, can be found in the guidance for therapeutic vapes.
Product standard MDSO 2024
The MDSO 2024 sets out the minimum safety and quality requirements for 'unapproved' therapeutic vaping devices and accessories.
The product design and safety requirements for ‘unapproved’ therapeutic vaping devices and accessories have been strengthened compared to the MDSO 2023 (refer to guidance on the MDSO 2023). The purpose of these product standards is to improve the quality, safety, and performance of the therapeutic vapes for patients that have a clinical need for these products.
MDSO 2024 requires:
- devices to be manufactured in a facility that has been certified to meet the quality management systems requirements that are specific to medical devices
- manufacturers to continually identify and mitigate risks related to the use of the device including risks or hazards that the TGA has prescribed
- ensuring access to accurate information about the design, manufacture and uses of these products
- the safety and performance risks be minimised by incorporating specific design and construction requirements
- manufacturers to implement designs that minimise the appeal of these products to youth and recreational users
- reinforcing that these products are therapeutic goods (as opposed to consumer goods).
- that the devices be subject to a toxicological risk assessment
Commencement and transitional provisions
From 1 March 2025 only unapproved therapeutic vaping devices and accessories that meet the MDSO 2024, or the EPs will be able to be imported into Australia (or manufactured in Australia).
It is expected that sponsors of existing therapeutic vaping devices or accessories will be required to re-notify to the TGA that their products are compliant against this standard or the EPs (as relevant).
From 1 July 2025, only those products that meet the MDSO 2024 or the EPs can be lawfully supplied in Australia.
The MDSO 2024 amends the MDSO 2023, and the following transitional provisions apply:
| Before 1 March 2025 | From 1 March 2025 to 1 July 2025 | From 1 July 2025 | |
|---|---|---|---|
| Imported into or manufactured in Australia | Comply with MDSO 2023 or MDSO 2024 | Comply with MDSO 2024 | Comply with MDSO 2024 |
| Supplied in Australia | Comply with MDSO 2023 or MDSO 2024 | Comply with MDSO 2023 or MDSO 2024 Stock rundown period | Comply with MDSO 2024 |
| Notification required | Existing sponsors may be required to notify compliance with MDSO 2024 (to continue to import after 1 March 2025) | For new sponsors, notify compliance with MDSO 2024 | For new sponsors, notify compliance with MDSO 2024 |
Requirements for unapproved therapeutic vaping devices and accessories under MDSO 2024
The requirements for MDSO 2024 can be broadly grouped into 8 categories:
- Quality management systems certification and compliance
- Risk management
- Plain device design and Pharmaceutical-like packaging
- Labels and Instructions for use
- Toxicological risk assessment
- Design and construction
- Electrical safety standards
- Battery safety standards
1. Quality management systems certification and compliance
(MDSO reference: See Schedule 1, Item 1)
A Quality Management System (QMS) is a formal and structured business system used for managing the quality of products and services. Medical device manufacturers use QMS for:
- the ongoing management of safety, performance, and quality of their devices
- keeping all documents as controlled and traceable documents
- meeting other regulatory requirements such as ongoing post-market requirements
- meeting customer requirements and expectations.
ISO 13485 is an important international standard that provides information on the necessary requirements for a QMS for manufacturers of medical devices. Manufacturers of therapeutic vaping devices and accessories must be certified as being compliant to the current version of ISO 13485. ISO 13485 certification, covering manufacturing of therapeutic vaping devices and accessories can be obtained from one of the following accredited bodies or organisations:
- an International Accreditation Forum (IAF) accredited organisation: a certification body accredited by a member of the IAF
- a notified body: an organisation recognised by the European Union to assess the conformity of medical devices before they can be marketed in the European Union
- an auditing organisation recognised under the Medical Device Single Audit Program (MDSAP)
Although MDSO 2023 allowed for certification to ISO 9001 as evidence of QMS compliance, from 1 March 2025 this will no longer be acceptable.
2. Risk management
(MDSO reference: See Schedule 1, Item 2)
Risk management for medical devices is the process of ensuring that risks related to the use of the device are reduced as far as possible. ISO 14971 is an important international standard that provides information on how to manage risks for medical devices. It is expected that the risk documentation you hold for therapeutic vaping devices and accessories will be in line with the ISO 14971 risk assessment process.
A risk management document is used in identifying, assessing, and minimising risk through the lifecycle of the device. The device lifecycle includes the design, manufacture, use, and disposal of medical devices. The risk considerations should cover scenarios where the device is used as intended by the manufacturer, as well as any foreseeable misuse or incorrect use. It is a living document to be kept current when there are changes to the design or manufacture of the device, or if there are new hazards identified. The hazards could be identified by the manufacturer’s experience or from complaints or adverse event reports. Changes to a device and the risk management document may also be necessary due to updates to regulatory requirements or standards.
The MDSO includes some of the risks that the manufacturer must reduce to an acceptable level:
- the toxicity of emissions from the therapeutic vaping device or accessory, including:
- the emissions over the expected lifetime of the device
- the materials used within the therapeutic vaping device or accessory, and
- any residues or contaminants.
For example:
- The assessment should consider substances that might leach into the inhalation pathway or from the device, chemicals that are emitted from the materials used in the device, and contaminants made by the heating element. This should include emissions of metals, volatile organic compounds, phthalates, pesticides, and nitrosamines
- Hazards or risks relating to batteries (if applicable), including the risk of fire or explosion.
For example:
- Batteries used in vaping devices that are discharged can become unstable, increasing the risk of overheating or explosion
- Vaping devices can overheat or catch fire, and this may cause battery explosions
- Button batteries may cause serious injury if swallowed
- Electrical hazards or risks
For example:
- Overcharging of batteries can cause the device to catch fire or explode
- Charging the device with equipment that does not meet electrical standards or is not compatible with the charger
- Risk of short-circuit while using or charging the device
- Using batteries that are not compatible with the vaping device, such as third-party batteries
- The usability of the therapeutic vaping device or accessory
For example:
- Controls or instructions that are unclear can lead to misuse of the device
- Foreseeable misuse of the therapeutic vaping device or accessory
For example:
- Adjusting wattage settings that may potentially increase the delivery of the vaping substance
- If the device uses original manufacturer pods, the use of third-party vaping substance pods, homemade pods, or tampering with substance pods
- Modification of the device that would lead to dangerous electrical or battery failure such as changing the battery to a non-approved battery.
- The containment and administration of a therapeutic vaping substance
For example:
- The intended dosage of the therapeutic vaping substance not being delivered
- Leakage from the device due to corrosion or cracks of the device material over time, particularly when exposed to heat or chemicals.
- Heating of the therapeutic vaping device or accessory
For example:
- Risk of burns from an overheating device (due to failure to adequately control the temperature of the atomiser of the device, for example)
- Overheating of the battery or electronic circuits of the device which may lead to failure of the device or harm to the user (e.g. burns)
3. Plain device design and pharmaceutical-like packaging
(MDSO reference: See Schedule 1, Items 10, 11)
Plain device design
Plain design requirements relate to the appearance of therapeutic vaping devices and accessories.
Restrictions on colours
A therapeutic vaping device or accessory must be mainly matte white or matte grey, and feature no more than 3 matte colours or shades including the colour or shade of any text.
This requirement is intended to ensure that the therapeutic vaping device or accessory has a plain appearance. If the device or accessory features a colour or shade other than matte white or matte grey, it must not be visible when the device is fully assembled. For example, you can use colours on internal components to help identify how to assemble the device, or to identify the therapeutic vaping substance to be used with the device or accessory.
How information must be displayed
Any information placed on a therapeutic vaping device or accessory must be:
- in English
- visible and clearly legible (e.g. the text must contrast with the colour of the device)
- durable
- in matte grey or matte black text, unless a relevant standard requires there to be to be another colour (for example, an internationally recognised warning symbol may be required to be yellow or red)
Prohibited display of claims and design elements
A therapeutic vaping device, or accessory must not:
- show any promotional statement, pictorial representation, or design
- show any words, symbols, trademarks, images, figures, logos, or emblems, which are inconsistent with another section of this standard
- include any features designed to change its appearance, including the following:
- heat activated inks
- inks or embellishments designed to appear gradually over time
- inks that appear fluorescent in certain light
- panels designed to be scratched or rubbed to reveal an image or text.
Pharmaceutical-like packaging
Pharmaceutical-like packaging requirements apply to the primary pack of a therapeutic vaping device or accessory. The primary pack is the complete package in which the vaping goods are supplied to customers. It is the visible pack sitting on the shelf that is sold through retail.
All therapeutic vaping devices and accessories must be designed and styled in a way that does not promote non-therapeutic use to consumers.
Restrictions on colours
The label and packaging of a therapeutic vaping device or accessory supplied in a primary pack must be white.
Information that must be included on the label of a primary pack
The label of a primary pack containing the therapeutic vaping device or accessory, at a minimum, must include the following warning statements:
- “KEEP OUT OF REACH OF CHILDREN”.
- “RISK OF FIRE EXPLOSION. REPLACE ONLY WITH SAME SIZE AND TYPE BATTERY”
- “WARNING CONTAINS BUTTON OR COIN BATTERY. HAZARDOUS IF SWALLOWED – SEE INSTRUCTIONS” - if applicable
Please note that the warning statement in point C is required only if the therapeutic vaping device or accessory contains a button or coin battery. More information can be found at Button and coin batteries | Product Safety Australia.
How information must be displayed
Information on the primary pack and the label of a primary pack of therapeutic vaping devices or accessories must:
- be in English
- be clearly legible
- be durable
- be in a text colour that is matte grey or matte black, unless a relevant standard requires there to be to be another colour (for example, an internationally recognised warning symbol may be required to be yellow or red)
- include warning statements that are:
- at least 5 mm in text size
- in bold-face, sans serif, capital letters of uniform thickness and size.
Prohibited display of claims and design elements
The primary pack or label of a primary pack containing the therapeutic vaping device or accessory must not:
- suggest that the device has health benefits other than its intended purpose, including healing, vitalising, natural, organic, or rejuvenating properties
- show any promotional statement, pictorial representation, or design
- show any words, symbols, trademarks, images, figures, logos, or emblems, which are inconsistent with another section of this standard
- include any features designed to change its appearance, including the following:
- heat activated inks
- inks or embellishments designed to appear gradually over time
- inks that appear fluorescent in certain light
- panels designed to be scratched or rubbed to reveal an image or text
- removable tabs
- fold out panels.
4. Labels and Instructions for use
(MDSO reference: See Schedule 1, Items 6, 7, 8, 9)
It is important to provide clear and accurate product labels and information with the therapeutic vaping device or accessory to communicate its identification and intended use, ensuring that it is not marketed in a way that appeals to youth. This allows health practitioners and individuals to make informed decisions regarding its use and treatment options.
Naming restrictions
To prevent a therapeutic vaping device or accessory from being marketed or designed in a way that is attractive to youth, we have restricted how they are named and prohibited the inclusion of any promotional material.
The name of a therapeutic vaping device or accessory must not:
- suggest that the device:
- has health benefits, including healing, vitalising, natural, organic, or rejuvenating properties, or
- is safe, without harm or without side effects
- be in any way attractive to children or adolescents
- promote the use or supply of the device
- exaggerate, or be likely to exaggerate, the efficacy or performance of the device
- encourage, or be likely to encourage, inappropriate or excessive use of the device
- indicate the device is associated with any food, beverage, or cosmetic product.
Compliance with these requirements will ensure that therapeutic vaping devices and accessories do not include names that are in any way promotional or designed to attract young people or new users, misleading, suggesting any health benefit, or resembling a food, beverage, or cosmetic product.
Labels
The label requirements for therapeutic vaping devices and accessories include information for patients that is intended to support identification of these products and are broadly consistent with requirements for other medical devices. Labelling requirements are intended to support identification of devices for patients in the event of an adverse event or product recall along with compliance and enforcement actions if required.
Information that must be included on the label of a therapeutic vaping device
The label placed on a therapeutic vaping device at a minimum must include:
- all the following information on the device itself:
- the sponsor’s name
- the device name, brand name, and model
- the batch code, lot number, or serial number that identifies the kind of medical device
- the warning statement: “Risk of fire or explosion. Replace only with the same size and type of battery”
- all the following information on the device itself, or on the packaging of the device if it is not possible to be on the device itself:
- sponsor’s address
- manufacturer’s name and address.
Information that must be included on the label of a therapeutic vaping device accessory
The label placed on a therapeutic vaping device accessory at a minimum must include:
- all the following information on the accessory itself:
- the device name and model
- the batch code, lot number, or serial number that identifies the kind of medical device
- all the following information on the accessory itself, or on the packaging or instructions for use of the device if it is not possible to be on the accessory itself:
- brand name
- sponsor’s name and address
- manufacturer’s name and address.
How labels should be displayed on therapeutic vaping devices and accessories
The label on the therapeutic vaping device and accessory must be either matte white, matte grey or matte black, unless a relevant standard requires it to be another colour, such as for warning symbols. All information on the labels must be:
- in English
- clearly legible
- durable
- in a text colour that is matte grey or matte black, unless a relevant standard requires there to be to be another colour (for example, an internationally recognised warning symbol may be required to be yellow or red)
- in a text size of (refer to Table 1):
- at least 3 mm for therapeutic vaping devices
- at least 1 mm for therapeutic vaping device accessories.
Instructions for use
These instructions for use (IFU) requirements broadly align with those of other medical devices and are intended to ensure that appropriate information about the design and intended use of therapeutic vaping devices and accessories is available to patients. The IFU also provides information to patients about how to use the device correctly, and any warnings and safety instructions that should be noted. These requirements will also support identification of these products in case of an adverse event. The IFU also includes information on end-of-life disposal of the device and its batteries.
Information that must be included in the IFU?
The IFU provided with a therapeutic vaping device or accessory at a minimum must include the following:
- manufacturer’s name, or trading name, and physical address (not a post office box)
- intended purpose of the device or accessory, the intended user of the device, and the kind of patient on whom it is intended to be used
- the name of the device
- the batch code, lot number, or serial number that identifies the kind of medical device
- any particular handling or storage requirements applying to the device, including recommended storage conditions
- any warnings, restrictions, or precautions that may apply in relation to the use of the device, including a statement that informs the patient or user to seek advice from a healthcare professional if the device or accessory is not functioning correctly
- if applicable, information about the risks associated with button or coin batteries
- information about any actions that need to be performed prior to using the device, including any preparatory actions for first-time use
- information about how to use the device, including instructions for assembly
- any cleaning requirements for the device
- information about the replacement of the consumable components of the device during its expected lifetime, including the size and type of battery
- either the expiry date for the device or the expected lifetime of the device, indicated as duration of use after the first use or the number of actuations before being discarded
- any precautions a patient or user should take if there are risks associated with the disposal of the device, including instructions on how to dispose of the device or its parts or how to recycle used batteries.
Information that should be displayed in the IFU?
All information provided in the IFU must be:
- in English, and it may also be provided in any other language
- clearly legible
- at least 3 mm in text size
- supplied in paper or electronic format that is readily accessible to patients.
Where the IFU is supplied in an electronic format, a paper copy must be provided to the patient upon request and without charge.
Where the IFU is supplied in an electronic format, a paper copy must be provided to the patient upon request and without charge.
Table 1: Text size specifications
| Text size specifications | Minimum text size |
|---|---|
| Text size for Instructions for use | 3 mm |
| Text size for labels on therapeutic vaping devices | 3 mm |
| Text size for labels on therapeutic vaping device accessories | 1 mm |
| Text size for labels on primary pack | 5 mm for all warning statements |
5. Toxicological risk assessment
(MDSO reference: See Schedule 1, Items 2(a)(i)(ii), 12)
Manufacturers of therapeutic vaping devices and accessories must have evidence of a toxicological risk assessment for the emissions from their device and materials used in the device, covering the entire expected lifetime of the device or accessory. This means that the toxicological testing and assessment is expected to be updated with any design or production changes, or emergence of new risks.
This assessment is essential to demonstrate that toxicological risks have been identified and minimised. International standards including ISO 10993 and ISO 18562 have relevant information on how to undertake a toxicological evaluation. The toxicological risk assessment must be based on evidence and include, at minimum, the following:
- the atomiser – this is the part of the device responsible for heating or vaporising the therapeutic vaping substance
- materials of the device that contact the therapeutic vaping substance or vapour – any materials within the device that come into contact with the e-liquid or vapour must be assessed
- leachable substances, contaminants, and degradation products – this includes any contaminants or by-products released due to heating, such as metals, volatile organic compounds (VOCs), phthalates, or other harmful substances.
In line with ISO 14971, the toxicological risk assessment must be integrated into the manufacturer’s overall risk management process. This standard requires manufacturers to mitigate risks, as far as possible, including, but not limited to:
- inhalation toxicity, emissions from heating elements and materials in contact with the therapeutic vaping substance or vapour and exposure to leachable substances, contaminants and any harmful by-products released during use
- the identity and quantity of any potential emissions, including any worst-case scenarios (e.g. if emissions increase as the device ages), and consider the risks they pose to the patient or other persons in the vicinity of use
- Determining, based on scientific rationale, whether the levels are acceptable or pose a risk of harm to a patient, or other persons in the vicinity of use
- identifying and implementing risk controls to address these toxicological risks
- monitoring the effectiveness of the risk controls and ensure that they remain adequate in consideration of the toxicological risk assessment results.
6. Design and construction requirements
(MDSO reference: See Schedule 1, Item 5)
Manufacturers must integrate the following requirements in the design and construction of their therapeutic vaping devices and accessories as detailed in Item 5 of the MDSO 2024:
- Emitted mass or dose specification: Ensure that the therapeutic vaping device or accessory is designed to deliver the intended amount of medication consistently and accurately. This information must be specified in the product’s documentation, such as the instructions for use. These devices should consistently produce the specified emitted mass or dose when operated according to the manufacturer’s instructions. Testing and validation must be conducted to verify the medication delivered by the devices.
- Reliable venting mechanism: Ensure that the therapeutic vaping device or accessory is designed with a reliable venting mechanism to safely manage pressure build-up in the event of battery failure. The venting system should direct excess pressure away from the user to minimise the risk of injury or harm to the user. The effectiveness of the venting mechanism should be verified under all operating conditions for the device.
- Minimise inadvertent actuation: Ensure that the therapeutic vaping device or accessory incorporates design features that prevent accidental activation of the device. This may include mechanisms such as dual operation controls, interlocks, secure activation controls, or other safeguards to ensure the device operates only when intended. The effectiveness of these features must be validated to ensure they reduce the risk of unintentional use.
- Maximum allowable temperatures of external parts and surfaces: Ensure that all external parts and surfaces of the therapeutic vaping device or accessory, which users may touch or handle (apart from the mouthpiece), do not exceed 48 degrees Celsius during its normal operation. The device must also be designed and constructed in a way that ensures that the external parts of the mouthpiece do not exceed 55 degrees Celsius during its normal use. The choice of materials and design features should be carefully considered to maintain these temperature limits and prevent potential harm to users.
- Durability: The therapeutic vaping device or accessory must be designed and constructed to withstand regular use and potential misuse or abuse. The device should remain intact and safe under stress conditions such as drops, impacts, or other conditions of stress. The device should not break, leak, or fail in a manner that could pose a safety risk to its user.
- Child-resistant features: Ensure that the therapeutic vaping device or accessory integrates child-resistant features to prevent children from operating the device or ingesting any hazardous components or substances. Features such as secure closures or compartments may be considered to reduce the risk of accidental ingestion or operation by children.
- Battery safety: For therapeutic vaping devices or accessories containing batteries, manufacturers must ensure compliance with relevant electrical safety standards including but not limited to those mentioned in this guidance (refer to ‘7. Electrical safety standards’). The manufacturer must design and construct their devices or accessories in a way that prevents overheating, short-circuits, and explosion risks during both use and recharging. This may include, but is not limited to, consideration of implementing appropriate battery protection circuits and safety measures including compliance with relevant standards in this guidance (refer to ‘8. Battery safety standards’) to address these risks.
- Leak prevention: Ensure that the therapeutic vaping device or accessory is designed in a way that prevents leaks of the therapeutic vaping substance being vaporized.
- Biocompatibility and material safety: Ensure all materials used in the therapeutic vaping device or therapeutic vaping device accessory are biocompatible and the level of exposure to harmful chemicals, such as heavy metals, VOCs, and other toxic substances, is below the level of detectable risk to human health. Manufacturers are expected to conduct toxicological risk assessments for the emissions from their device and materials used in the device, covering the expected lifetime of the therapeutic vaping device or accessory as part of their risk management process (refer to section ‘5. Toxicological risk assessment’ of this guidance).
These design and construction requirements focus on safety and performance risks associated with therapeutic vaping devices and accessories, including accurate dosage delivery, safe temperatures of the external contact points of the devices, preventing accidental activation, battery safety, usage-related risks, and child safety features.
7. Electrical safety standards
(MDSO reference: See Schedule 1, Items 2(a)(iv), 4, 5(g))
Manufacturers of therapeutic vaping devices and accessories must ensure compliance with the following Australia and New Zealand electrical safety standards to ensure the safe operation of their devices.
If your therapeutic vaping device or accessory operates electrically, it must meet the following standards:
- AS/NZS 3820 – this standard outlines essential safety requirements for low-voltage electrical equipment
- AS/NZS 4417.1 – this standard requires proper marking of electrical products to indicate compliance with relevant regulations.
8. Battery safety standards
(MDSO reference: See Schedule 1, Item 3)
Therapeutic vaping devices often use batteries that pose risks such as overheating, fires, and in the worst-case explosions.
To reduce these risks, manufacturers must comply with the following battery standards based on the battery type (e.g. lithium or nickel-based battery) that applies to their therapeutic vaping device or accessory:
- Button batteries: manufacturers must adhere to the following standards:
- the Consumer Goods (Button/Coin Batteries) Information Standard 2020
- the Consumer Goods (Button/Coin Batteries) Safety Standard 2020
- the Consumer Goods (Products Containing Button/Coin Batteries) Safety Standard 2020
- the Consumer Goods (Products Containing Button/Coin Batteries) Information Standard 2020
More information on compliance against these standards can be found at Button and coin batteries | Product Safety Australia.
- Non-button primary batteries: manufacturers are required to have their device certified to IEC 62133-1 from an accredited certification body to demonstrate that the battery complies with relevant parts (Parts 1, 2 and 4 or 5) of that standard.
- Lithium secondary batteries: manufacturers must have:
- UN/DOT 38.3 certification, or a declaration of conformity with UN/DOT 38.3, supported by independent test reports from an ISO 17025 accredited laboratory, and
- IEC 62133-2 certification from an accredited certification body.
- Nickel secondary batteries: manufacturers are required to have IEC 62133-1 certification from an accredited certification body to demonstrate that the battery complies with that standard.
Post-market requirements
We may ask for you for evidence that you comply with the regulatory requirements. Upon request, sponsors will be required to provide such evidence within a specified time frame, which will be at least 5 working days.
Appendix A
The following excerpt outlines the exemption conditions for therapeutic vaping devices and accessories under item 2.17 of Schedule 4 of the MD Regulations, applying specifically to those using the unapproved pathway:
(a) The sponsor must give the Secretary a notice (the sponsor notice), in a form approved in writing by the Secretary, stating that:
(i) the device is intended, by the person under whose name the device is or is to be supplied, only to administer or contain a therapeutic vaping substance whose only indications are use for smoking cessation or the management of nicotine dependence; and
(ii) the device complies with the Essential Principles or is imported or supplied (as the case may be) with the consent of the Secretary under section 41MA or 41MAA of the Act.
(b) The sponsor notice must be given as follows:
(i) for a device imported into Australia on or after 1 March 2024—before the device is imported;
(ii) for a device imported into Australia before 1 March 2024—before the earlier of the time the device is supplied to the ultimate consumer and the end of the period of 2 months beginning on the day Schedule 1 to the Therapeutic Goods Legislation Amendment (Vaping Reforms) Regulations 2024 commences;
(iii) for a device manufactured in Australia on or after 1 March 2024—before the device is first supplied in Australia;
(iv) for a device manufactured in Australia before 1 March 2024—before the earlier of the time the device is supplied to the ultimate consumer and the end of the period of 2 months beginning on the day Schedule 1 to the Therapeutic Goods Legislation Amendment (Vaping Reforms) Regulations 2024 commences.
(c) The sponsor holds information or evidence to support the statements made in the sponsor notice.
(d) Neither of the statements made in the sponsor notice is incorrect.
(e) The device is not the subject of a determination, by the Secretary and published on the Department’s website, that the supply of the device be stopped or should cease because:
(i) the Secretary is satisfied that the supply compromises public health and safety; or
(ii) the Secretary is satisfied that the device does not comply with the Essential Principles.
(f) The sponsor must:
(i) if requested by the Secretary, give the Secretary the information or evidence referred to in paragraph (c); and
(ii) do so within the period requested by the Secretary (which must be at least 5 working days starting on the day on which the Secretary’s request is made).
(fa) The sponsor must:
(i) if requested by the Secretary, give the Secretary a reasonable number of samples of the device; and
(ii) do so within the period requested by the Secretary (which must be at least 5 working days starting on the day on which the Secretary’s request is made).
(fb) The sponsor must allow an authorised officer:
(i) to enter, at any reasonable time, any premises (including premises outside Australia) at which the sponsor or any other person deals with the device; and
(ii) while on those premises, to inspect those premises and the device and to examine, take measurements of, conduct tests on, require tests to be conducted on or take samples of the device or any thing on those premises that relates to the device; and
(iii) while on those premises, to make any still or moving image or any recording of those premises or any thing on those premises.
(fc) The sponsor must, if requested to do so by an authorised officer, produce to the authorised officer such documents relating to the device as the authorised officer requires and allow the authorised officer to copy the documents.
(fd) If the sponsor is not the manufacturer of the device, the sponsor must have procedures in place to ensure that the manufacturer of the device allows an authorised officer:
(i) to enter, at any reasonable time, any premises (including premises outside Australia) at which the manufacturer or any other person deals with the device; and
(ii) while on those premises, to inspect those premises and the device and to examine, take measurements of, conduct tests on, require tests to be conducted on or take samples of the device or any thing on those premises that relates to the device; and
(iii) while on those premises, to make any still or moving image or any recording of those premises or any thing on those premises.
(fe) If the sponsor is not the manufacturer of the device, the sponsor must have procedures in place to ensure that the manufacturer of the device, if the manufacturer is requested to do so by an authorised officer, produces to the authorised officer such documents relating to the device as the authorised officer requires and allow the authorised officer to copy the documents.
(g) The device may be supplied to a person who is not the ultimate consumer of the device only if:
(i) the person (the recipient) to whom the device is supplied is the holder of a licence in force under Part 3‑3 of the Act that authorises a step in the manufacture of vaping goods; or
(ii) the recipient is a wholesaler, pharmacist, medical practitioner or nurse practitioner who is the holder of a licence, or is otherwise authorised, to supply one or more substances included in Schedule 4 to the current Poisons Standard under a law of the State or Territory in which the recipient carries on a business, practises or is employed; or
(iii) the Secretary has given the recipient a consent under subsection 41RC(1) of the Act to supply the device; or
(iv) in the case of a device that is covered by a determination made by the Minister under section 41R of the Act—the recipient is specified in the determination, or is included in a class of persons specified in the determination, in relation to the device.
(h) The device may be supplied to the ultimate consumer of the device only if:
(i) the device is supplied as a finished product; and
(ii) the supply is by a pharmacist, medical practitioner or nurse practitioner who is the holder of a licence, or is otherwise authorised, to supply one or more substances included in Schedule 4 to the current Poisons Standard under a law of the State or Territory in which the recipient carries on a business, practises or is employed; and
(iii) if the supply is by a person authorised as described in subparagraph (ii), the supply is in accordance with that authorisation.
(i) The sponsor must:
(i) keep records relating to the source and supply of the device; and
(ii) if requested by the Secretary, give the records to the Secretary within the period requested by the Secretary (which must be at least 5 working days starting on the day on which the Secretary’s request is made).
(j) The sponsor must provide information of a kind mentioned in subsection 41MP(2) or 41MPA(2) of the Act relating to the device to the Secretary within the following periods:
(i) if the information relates to an event or other occurrence that represents a serious threat to public health—48 hours after the sponsor becomes aware of the event or occurrence;
(ii) if the information relates to an event or other occurrence that led to the death, or a serious deterioration in the state of health, of a patient, a user of the device, or another person—10 days after the sponsor becomes aware of the event or occurrence;
(iii) if the information relates to an event or other occurrence a recurrence of which might lead to the death, or a serious deterioration in the state of health, of a patient, a user of the device, or another person—30 days after the sponsor becomes aware of the event or occurrence;
(iv) in any other case—60 days after the sponsor becomes aware of the information.
Glossary
| Name | Description |
|---|---|
| Australian Register of Therapeutic Goods (ARTG) | the public database of therapeutic goods that may be lawfully supplied in Australia. The ARTG is the main pathway for consumers to access medicines, biologicals and medical devices in Australia. |
| Authorised Prescriber (AP) Scheme | allows authorised medical practitioners to prescribe therapeutic goods that are not included in the ARTG to certain patients with a particular medical condition. |
| Contaminant | a chemical that is not intended to be in a therapeutic vaping device or therapeutic vaping device accessory (or therapeutic vaping substance) |
| MD Regulations | Therapeutic Goods (Medical Devices) Regulations 2002. |
| Therapeutic vaping device | a therapeutic good that is a vaping device, other than: (a) a disposable therapeutic vape (within the meaning of the Therapeutic Goods Regulations 1990); or (b) a therapeutic cannabis vaping device. |
| Therapeutic vaping device accessory | means a therapeutic good that is an unfilled cartridge, capsule, pod or other vessel: (a) that is designed or intended for use in, or with, a therapeutic vaping device; and (b) that is designed or intended to contain a therapeutic vaping substance; and (c) whether or not the cartridge, capsule, pod or other vessel is designed or intended to be refilled; but does not include a therapeutic cannabis vaping device accessory. |
| Therapeutic vaping kit | means a kit, covered by subsection 7B(1) of the Act, that: (a) contains one or more therapeutic vaping substances or therapeutic vaping substance accessories; and (b) does not contain any other goods. |
| Therapeutic vaping pack | means a primary pack that: (a) contains at least one therapeutic vaping substance or therapeutic vaping substance accessory; and (b) contains at least one therapeutic vaping device or therapeutic vaping device accessory; and (c) does not contain any other therapeutic goods. |
| Therapeutic vaping substance | means a therapeutic good that is a liquid or other substance designed or intended for use in or with a vaping device. |
| Therapeutic vaping substance accessory | means a vaping accessory that: (a) is designed or intended for use in, or with, a therapeutic vaping device; and (b) contains a therapeutic vaping substance. |
| Therapeutic vape | expression used in this guidance for convenience to refer to a therapeutic vaping device, accessory, substance, or substance accessory. |
| TG Act: | Therapeutic Goods Act 1989 (Cth). |
| TG Regulations | Therapeutic Goods Regulations 1990 (Cth). |
| Unapproved therapeutic goods | therapeutic goods that are not included in the ARTG and have not been evaluated by the TGA for quality, safety and efficacy. There are established pathways under the TG Act that allow access to unapproved therapeutic goods in certain circumstances, including the Special Access Scheme and the AP Scheme. |