This page was updated on [date_placeholder]. See page history for details.
Purpose
The purpose of this guidance is to assist sponsors and manufacturers of spinal implantable medical devices to understand and comply with new regulatory requirements.
From 25 November 2021, some spinal implantable medical devices will be required to be reclassified.
The new regulatory requirements will also include:
more detailed assessment of the manufacturer’s quality management systems and assessment of technical documentation related to each device
conformity assessment documents demonstrating procedures appropriate for their classification
a mandatory audit assessment by the TGA for device inclusion applications, including assessment of clinical evidence
Class IIb spinal fusion devices will need to have specific information in the ARTG entry about the devices that are supplied under the ARTG entry. This includes product names of all devices under each ARTG entry.
The classification of your spinal implantable medical device will determine the safety and performance requirements to be demonstrated to meet regulatory requirements.
Spinal implantable medical devices are used in different parts of the spine (cervical, thoracic, lumbar and pelvic) to address a number of health problems including arthritis of the spine, deformities of the spine (including scoliosis – abnormal curvature of the spine), spondylolisthesis, herniated discus, trauma, spinal tumours, etc.
Spinal implantable medical devices include the following:
motion-preserving devices for the spine
implantable devices that come into contact with the spinal column.
On 12 December 2019, the Regulations were amended in relation to a number of medical devices, including the classification of some spinal implantable medical devices.
The amendments include the reclassification of motion-preserving devices (such as spinal disc replacements and devices used to help restore the normal motion of the spine) and spinal implantable devices that come into direct contact with the spinal column to Class III, effective from 25 November 2021.
These changes are part of a broader range of medical device reforms to align the Regulations with the European Union (EU) framework, following two rounds of public consultation.
Further regulatory refinements were made on 29 October 2021 to provide greater clarity around the regulation of these products.
Devices being reclassified from Class IIb to Class III
Implantable devices that are intended to be a motion-preserving device for the spine will be reclassified from Class IIb to Class III.
From 25 November 2021, the following classification rule will apply:
(4B) If the device is intended by the manufacturer to be a motion-preserving device for the spine (such as a spinal disc replacement), the device is classified as Class III.
Examples of devices being reclassified to Class III
Devices that articulate and preserve the motion of the spine will be reclassified to Class III.
Ancillary devices, such as screws, plates, hooks and rods intended for use during spinal fusion procedures, and in procedures preserving mobility, remain Class IIb, as long as they are not explicitly used in preserving the motion of the spine.
Spinal disc replacement implants – not intended to fuse vertebral bodies
Artificial disc replacements are examples of spinal implants used in non-fusion procedures.
Total disc replacement is a surgical procedure in which a diseased or damaged intervertebral disc is replaced with an artificial disc to restore the normal function and movement of the spine.
The image shows a spinal disc replacement implant. The device has a circular design with a white or cream main body in two components, connected by a grey coloured joint. Extending from the perimeter of the main body are several yellow, components or protrusions.
A technology that helps to restore normal spinal motion at the involved levels.
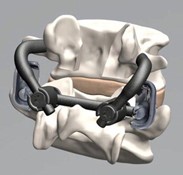
The AFRS consists of a precision instrumentation set and an anatomic facet implant family whose design is based upon a comprehensive computed tomographic morphology study of the facet joint.
The system utilises traditional pedicle screw fixation of its superior and inferior facet implants.
Image of an Anatomic facet replacement system (AFRS)
The image depicts an anatomic facet replacement system (AFRS) medical device. It consists of two white prosthetic components connected by a dark metal joint.
Interspinous process spacer
The device aims to stabilize the motion segments by preserving natural mobility.
The implant is comprised of a spacer designed to be implanted between two lumbar spinous processes.
The image shows a line drawing or illustration of an interspinous process spacer medical device.
The device appears to be composed of interconnected mostly white and one blue component.
The image shows how the components work together to provide spinal stabilisation and mobility,
Spinal implantable devices will remain Class IIb
Spinal implantable devices, such as screws, cages, plates, hooks, or rods that are not intended to be used as a motion-preserving device will remain classified as Class IIb.
Devices that will remain Class IIb will be subject to mandatory audit.
Examples of spinal implantable devices:
screws
cages (static and expandable)
plates
hooks
rods
bone graft/orthopaedic cement*
cables/wires
*Some bone graft/orthopaedic cement may be Class III, depending on where it is intended to be used and what materials it contains (e.g. medicine/material of animal or microbial origin).
What you need to do
If you are a sponsor of a spinal implantable medical device, the action you will need to take to comply with the new regulations will depend on the status of your product.
Medical devices included in the ARTG prior to 25 November 2021
Applications to include a medical device in the ARTG lodged before 25 November 2021
Applications to include a new medical device in the ARTG on or after 25 November 2021
Medical devices included in the ARTG before 25 November 2021
If you have a Class IIb medical device inclusion in the ARTG with a start date before 25 November 2021, transitional arrangements are in place to ensure that you can continue to supply your device while you apply for it to be included in the ARTG as a Class III medical device.
To continue to supply your device you must:
Notify the TGA before 25 May 2022 that you have an inclusion that will need to be reclassified
Submit a reclass application for your device to be included in the ARTG as a Class III medical device before the transition deadline.
If you do not intend to continue supplying the device, you should cancel your ARTG inclusion before 25 May 2022.
If you notify the TGA of your devices before 25 May 2022 but you do not submit an application for a Class III inclusion before the transition deadline, you must cease supplying your device from the day of the transition deadline and cancel your inclusion.
Applications to include a medical device in the ARTG lodged before 25 November 2021
If you have submitted an application for inclusion in the ARTG for a Class IIb device before 25 November 2021, your application will be assessed, and the device will be included in the ARTG as a Class IIb device under the old classification rules.
To be eligible for the transitional arrangements to reclassify your device as a Class III device, you must:
Notify the TGA that you have an ARTG inclusion that will need to be reclassified by whichever is the later date:
before 25 May 2022
within 2 months of the start date of your ARTG entry
Submit a reclass application for your device to be included in the ARTG as a Class III device before the transition deadline.
Cancelling your ARTG inclusion
If you do not notify the TGA before 25 May 2022 or within two months of the start date for your ARTG entry (whichever is the later date) of your intention to apply for the device to be included in the ARTG as a Class III device you will no longer be eligible for the transitional arrangements. You should:
cease supply of your device from 25 May 2022
cancel your ARTG inclusion before 25 May 2022.
If you notify the TGA of your device before the due date, but you do not submit an application for a Class III ARTG inclusion before the transition deadline, you must:
cease supply of your device from the day of the transition deadline
cancel your ARTG inclusion.
Applications to include a new medical device on or after 25 November 2021
Any application for inclusion of a new device intended to be a motion-preserving device for the spine that is not yet included in the ARTG, submitted to the TGA on or after 25 November 2021 must be submitted as an application for a Class III medical device.
You must use the Class III Device Application form in TGA Business Services. If you submit your application using any other form (e.g. another classification form) your application will fail and your application fee will not be refunded to you.
Notifying the TGA that you need to reclassify a device
To notify the TGA about an ARTG inclusion for a Class IIb spinal implantable medical device that needs to be reclassified, you will need to fill in the online form on our Consultation Hub:
The form will be available until 24 May 2022.
The information you will need to provide includes the existing ARTG number, current classification, and new classification. For devices that will be newly classified as Class III medical devices, you will also need to provide UPIs for each device and/or variant.
Reclassifying an ARTG inclusion as a Class III device
Kind of device: Class IIb vs Class III
The requirements you will need to meet when submitting an application for inclusion of a Class III device in the ARTG are different from the requirements for Class IIb devices.
Only devices of the same kind can be supplied under one ARTG entry.
Class IIb devices that share the following characteristics are of the same “kind” and can be included in one ARTG entry:
supplied by the same sponsor
manufactured by the same manufacturer
have the same classification
have the same Global Medical Device Nomenclature (GMDN) System Code
Class III devices must have the same Unique Product Identifier (UPI) in order to be considered of the same “kind”.
The devices included in your current Class IIb ARTG entry may have different UPIs.
You will require a separate Class III application for inclusion in the ARTG for each UPI.
A Class III ARTG entry may still cover more than one device, but only if the
devices have the same UPI
variation in the design of the devices is only to accommodate different patient anatomical requirements
variation does not change the intended purpose of the device
Timeframes for ARTG inclusion applications as Class III medical devices
To continue supplying your devices, you must submit your reclassification application for a Class III inclusion in the ARTG before the transition deadline.
In November 2023, regulatory amendments have been made to extend the transition deadline for some transitional devices.
The updated transitional deadline for spinal implantable medical devices (motion preserving) is 1 July 2029.
More information about the TGA’s strategy in response to EU MDR transition extension can be found on our EU MDR transition extension page.
If you have submitted your application before this date, but it has not yet been finalised by the TGA, you are able to continue to supply your devices using your Class IIb ARTG entry until a decision is made about your Class III application.
How to submit a reclassification application
Create a ‘New Device Application’ from the menu in the eBS Portal
Select "Medical Device - Included" from the first drop down list provided
Select the option to ‘Reclassify an existing register entry’
Search for the ARTG Number to be reclassified: eg. 130099 (example only)
Select the "Clone" button.
Allow the system to clone the information associated with the ARTG entry into the application.
Select Class III from the drop down provided for the “New classification” question
If the GMDN code in the existing entry has been made obsolete or has been updated, the sponsor is responsible for selecting the most appropriate and current code available in the GMDN agency database.
If you are required to select a new GMDN code that is different to the cloned ARTG entry, you will not be able to validate and submit the application.
Save the draft application and email TGA Devices info line at devices@tga.gov.au for assistance.
If there is a change of manufacturer, you must submit a new application (i.e. select “Create a new inclusion in the register” instead of “Reclassify an existing register entry” in the Step 3 shown above) and provide information about the existing ARTG entry in the application form or in a supporting document attached with the form.
What to include in your application
Class III applications for ARTG inclusion must be accompanied by appropriate Conformity Assessment documentation in order to pass preliminary assessment.
The required documents are outlined in the final column "Documentation to be provided with the application (Evidence of product assessment)" of Table 2 in the Use of market authorisation evidence from comparable overseas regulators / assessment bodies for medical devices (including IVDs).
The applicable product assessment evidence (e.g. Design Examination Certificate) must be provided in addition to the Manufacturer Evidence.
A one-page document attached to your application, outlining the intended purpose of the device and whether the device is for spinal fusion or motion preserving will help assessors to process your application.
Ensure you allow sufficient time to obtain your conformity assessment documentation to submit your documents with your application.
If the conformity assessment documentation is not sufficient, the application cannot pass preliminary assessment, your application will be refused, and you will not be able to transition your device to the new classification
Mandatory audits
If your Class III application is not supported by MDR, TGA or AU CAB certification, your application will be selected for a compulsory application audit.
Compulsory application audits attract an audit assessment fee and require submission of additional information, which may include, clinical evidence to support the safety and performance of the device.
Further, Class IIb spinal implantable devices that are not supported by MDR, TGA or AU CAB certification will also need to undergo compulsory application audit from 25 November 2021. These audits may include assessment of clinical evidence.
The Clinical Evidence Guidelines outline the level of evidence expected for medical devices to be included in the ARTG.
You will need to pay the relevant application fee and audit assessment fee.
You can request abridgement and reduction of the assessment fee of the audit assessment (including requests to abridge the level of audit if appropriate).
Class IIb applications/entries
The requirements you will need to meet when submitting an application for inclusion of a Class IIb spinal implantable device in the ARTG are different from the general requirements for Class IIb devices.
A one-page document attached to your application, outlining the intended purpose of the device and whether the device is for spinal fusion or motion preserving will help assessors to process your application.
Devices supplied under the ARTG entry
Under Regulation 5.12, from 25 November 2021, Class IIb spinal fusion devices will need to have specific information in the ARTG entry about the devices that are supplied under the ARTG entry.
This includes product names of all devices under each ARTG entry, before those devices can be imported, supplied or exported.
For new applications, Class IIb application forms have been updated to provide space to include this information.
Sponsors of any Class IIb spinal fusion devices that are currently in the ARTG, or will be prior to 25 November 2021, will need to notify the TGA of the product names.
Sponsors are encouraged to provide this information to the TGA as soon as possible in order to allow time for ARTG entries to be updated.
Notifying the TGA: Class IIb spinal fusion devices
Sponsors with Class IIb spinal fusion devices were required to notify the TGA before 25 November 2021 and update the entry with the current product names.
A temporary form, ‘Class IIb Product Name Variation Form’ was made available in the TBS portal for sponsors to notify the TGA and update their entry per this requirement.
This form has now been removed from the TBS portal and is no longer available for use.
If you wish to amend, remove or add product names in your Class IIb entry you should submit a Device Change Request (DCR) through your TBS portal.
However, if your spinal implantable medical device is a screw, wedge, plate, wire, pin, clip, connector, or similar article then you may be excluded from the obligation to provide the patient information materials with the device.
Amendments to Prostheses List
Some surgically implantable prostheses and other eligible devices are listed on the Prostheses List.
The Prostheses List contains information about the device, including the ARTG entry.
If your Class III ARTG inclusion application with the TGA is successful and a new ARTG number is assigned, you should apply for your Prostheses List billing code to be amended to ensure the information entered on the Prostheses List is accurate.
Failure to maintain the currency of your Prostheses List billing codes’ details may result in discrepancies between private hospital records and the Prostheses List and consequently may cause delays and inaccuracies in benefits paid by private health insurers.
To update the Prostheses List with your new ARTG numbers, you will need to submit an Amendment Application using the Prostheses List Management System (PLMS) (this system is only accessible for registered users of the system) for each of the affected billing codes.
When submitting an Amendment Application, ensure you clearly explain the reason for the change and include relevant supporting documentation.
Examples of supporting documentation include:
A new ARTG certificate
A catalogue or product brochure that provide relevant information about the devices affected by the change
For any further queries about Prostheses List, contact the Department via email: prostheses@health.gov.au
If your inclusion application is not successful
If your inclusion application to transition your device to the new classification is not successful, you will be notified of the decision in writing and you will be provided the reasons for the decision.
If you are not satisfied with this decision, you may request reconsideration of this initial decision under section 60 of the Therapeutic Goods Act 1989 within 90 days of the decision.
If you are not satisfied with the reconsideration (reviewable decision), you may apply to the Administrative Appeals Tribunal or the court.
When you must cease supply using your old ARTG entry
If you do not meet your obligations under the transitional arrangements, you will need to cease supply of your devices that would be considered Class III medical devices.
The following table outlines the circumstances and timeframes:
If...
You must...
You have not notified the TGA that your device needs to be reclassified before 25 May 2022, or within two months of inclusion of your device under the old classification rules (whichever is the later date)
Cease supply of your devices from 25 May 2022 or the date that is 2 months after the start date of your ARTG entry (whichever is the later date).
You have not submitted an application for inclusion in the ARTG to transition your device to the correct classification before the transition deadline
Cease supply of your devices from the day of the transition deadline.
Your application for ARTG inclusion of your device with the correct classification is unsuccessful.
Cease supply of your device from the time that you are notified of the outcome of your application.

{kind=link}
{kind=link}
{kind=link}
{kind=link}




