Preparing for technical file review (application audit) for in-vitro diagnostic medical devices
Guidance for preparing a technical file review, as part of your application to include an in-vitro medical device (IVD) in the Australian Register of Therapeutic Goods.
Purpose
The Therapeutic Goods Act 1989 (the Act) and Therapeutic Goods (Medical Devices) Regulations 2002 (the Regulations) specify that:
- applications to include certain IVD medical devices (IVDs) in the ARTG must be selected for an application audit - an application audit assessment fee will be charged
- the TGA may also select any other application for inclusion for an application audit - an audit assessment fee will not be charged for these audits.
For an IVD, an application audit is primarily a review of the manufacturer's technical documentation and will often be referred to as a technical file review (TFR)1.
When conducting an application audit, the TGA may ask for any information or documents which may be relevant to demonstrating compliance with the Essential Principles for safety and performance, the conformity assessment procedure which has been applied by the manufacturer or information about advertising or supply of the IVD.
If an application audit is to be conducted, we will write to the sponsor who submitted the application to include the IVD on the ARTG advising:
- that the application has been selected to undergo an application audit;
- the documentation that the TGA requires the sponsor to provide;
- if applicable, the fee that is payable. The TGA will send a separate invoice formally requesting the payment - the invoice will provide the payment options and the due date for payment.
It should be noted that sponsors cannot submit the documentation before we formally writing to request it.
What an application audit involves
Section 41FI of the Act specifies that there are two aspects of an application that the TGA can consider when conducting an application audit:
- whether the application complies with the requirements of the Act and the Regulations; and
- whether matters that the sponsor has certified in submitting the application are correct.
Examples of what the TGA will consider when conducting an application audit are:
- Is the product a medical device as defined by section 41BD of the Act?
- Is the product an IVD medical device as defined by Regulation 1.3?
- Are the variant details and Unique Product Identifier (UPI) details valid in the application, where applicable?
- Is the GMDN term in the device application appropriate for the device?
- Based on the manufacturer's intended purpose, the details in the application form, and the information provided by the sponsor, has the device been correctly classified in the Australian Declaration of Conformity and the device application?
- Is there any evidence of non-compliance with any of the Essential Principles in Schedule 1 of the Regulations?
- Is the manufacturer's Australian Declaration of Conformity in compliance with the requirements of Schedule 3 of the Regulations?
- Is the conformity assessment procedure appropriate for the classification of the device?
- Have representative labelling and Instructions for Use been provided, and do they demonstrate compliance with Essential Principle 13?
- Has a risk management report been included and is it applicable to the IVD medical device?
- Does the stability data support the stability claims?
- Does the clinical evidence meet the requirements of:
- Essential Principle 14, Schedule 1 of the Regulations
- Part 8, Schedule 3 of the Regulations?
- Does the analytical performance data meet the requirements of Essential Principle 15, Schedule 1 of the Regulations?
Technical file
A technical file represents all the information that is held by a manufacturer in relation to a particular IVD.
The documentation is normally an output of the manufacturer's quality management system, and includes information generated throughout the design, development, production and monitoring phases of the IVD.
Information may be held across a number of locations or in different forms, and various components from the technical file can be used to demonstrate conformity to the Essential Principles of safety and performance.
The Global Harmonisation Taskforce (GHTF) has published the Summary Technical Documentation for Demonstrating Conformity to the Essential Principles of Safety and Performance of In Vitro Diagnostic Medical Devices (STED) as guidance on the relevant documentation that should be assembled from the manufacturer's existing product technical file for submission to a regulatory body (such as the TGA) for pre-market approval.
The selected documentation is referred to as a STED (Summary Technical Documentation).
While it is not mandatory for manufacturers to adhere to all the recommendations outlined in the GHTF STED document, it does provide useful guidance for identifying documentation that may be requested by the TGA as part of a TFR.
We prefer that the information be submitted for application audits in the STED format.
The depth and detail of the information contained in the STED is primarily dependent on the risk classification of the IVD, however consideration should also be given to the complexity of the IVD and whether it incorporates the detection or measurement of a new analyte, any new technology or a new clinical application.
Application audit process
The following flowchart summarises the process for an application audit:
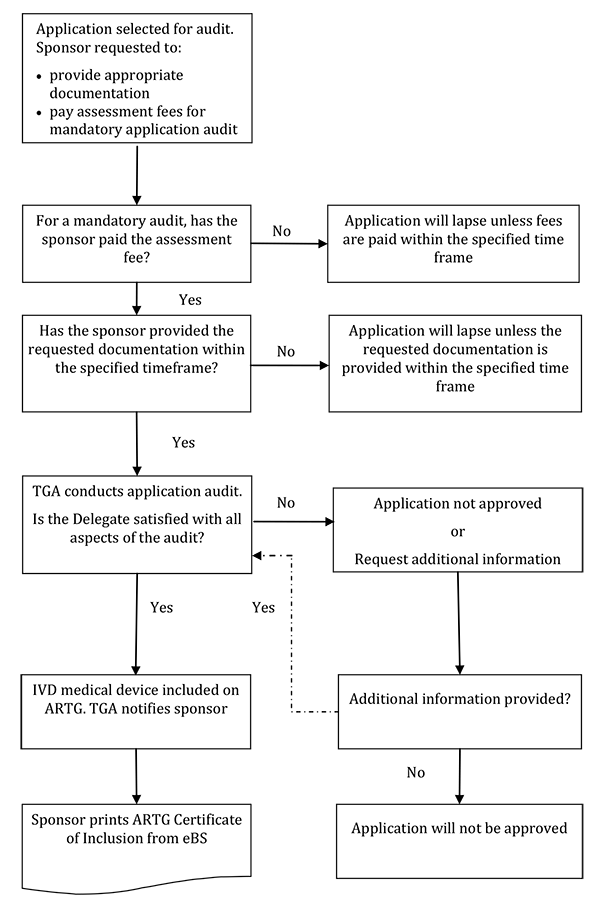
{kind=link}
This text representation of this flowchart is provided as a list with numbered steps.
- Application selected for audit. Sponsor requested to:
- provide appropriate documentation
- pay assessment fees for mandatory application audit
- For a mandatory audit, has the sponsor paid the assessment fee?
- No: Application will lapse unless fees are paid within the specified time frame - end flowchart
- Yes: Go to Step 3
- Has the sponsor provided the requested documentation within the specified timeframe?
- No: Application will lapse unless the requested documentation is provided within the specified timeframe - end flowchart
- Yes: Go to Step 4
- TGA conducts application audit. Is the Delegate satisfied with all aspects of the audit?
- No: Application not approved (end flowchart) OR Request additional information
- Additional information provided?
- No: Application will not be approved - end flowchart
- Yes: Return to Step 4
- Additional information provided?
- No: Application not approved (end flowchart) OR Request additional information
- Yes: IVD medical device included on ARTG. TGA notifies sponsor. Go to Step 6
- Sponsor prints ARTG Certificate of Inclusion from eBS - end flowchart
The possible outcomes of an application audit are:
| If the audit | then | and |
|---|---|---|
| is successful and the sponsor has paid the appropriate fees | we will notify the sponsor that the application for inclusion in the ARTG has been successful | the sponsor can print the Certificate of Inclusion from eBS |
| lapses | the sponsor will need to re-apply to include the device in the ARTG | pay any fees associated with a new application |
| is not successful | we will notify the sponsor that the application has not been successful and the reasons for the decision | the sponsor should ensure that they have addressed any deficiencies in the information provided to the TGA before they re-apply to include the device in the ARTG |
Applications that are subject to a mandatory application audit
Regulation 5.3 specifies the IVDs that must be selected for an application audit. However, where the IVD is included in the scope of a current Conformity Assessment Certificate issued by the TGA, or the device is an in-house IVD, an application audit is not required.
The following devices must be selected for an application audit:
- non assay-specific quality control material that is intended for monitoring a Class 4 IVD
- an IVD that is intended for self testing
- an IVD that is intended for point of care testing
- a Class 3 IVD that is intended for detecting the presence of, or exposure to, a sexually transmitted agent
- an IVD for managing and monitoring the treatment of infections diagnosed using a Class 4 IVD medical device (for example, quantitative nucleic acid test (NAT) and genotyping assays for HIV and HCV)
- an IVD that is intended to be supplied for use under the pharmaceutical benefit scheme
- an IVD that is intended to be supplied for use in a national screening program
- an IVD for which the TGA is not satisfied that the body or authority that issued the manufacturer's evidence has appropriate authority and expertise to assess the IVD against the Australian conformity assessment procedures. For example, a Class 3 IVD that is supported by an ISO 13485 certificate and does not also have a Class III or Class IV Medical Device Active Licence Listing issued by Health Canada-in this case, evidence of appropriate product assessment has not been provided.
Applications selected for non-mandatory audit
We may select any application which is not subject to a mandatory audit under Regulation 5.3 for a non-mandatory application audit - an audit assessment fee will not be charged for these audits.
Non-mandatory audits may be undertaken to verify that the application has been made in accordance with section 41FC and that the matters certified under section 41FD are correct.
We will write to the sponsor requesting submission of the information required to conduct the application audit.
Application audit assessment fees
An assessment fee is payable for each mandatory application audit that is required by the therapeutic goods legislation.
Where an application includes more than one IVD that is subject to a mandatory audit, an assessment fee is payable for the first audit, however no fee will be charged for any additional audits conducted under the same application.
Fees are not payable for non-mandatory application audits conducted by the TGA.
If multiple applications for inclusion in the ARTG are submitted for IVDs requiring mandatory application audit, and the technical documentation shared between these IVDs is sufficiently similar, then a reduction in assessment fees will be considered, provided that:
- effective applications are received by the TGA on the same day (i.e. the application fees are paid on the same day); and
- the applications are submitted by the same sponsor;
- the IVDs have the same manufacturer; and
- the documentation required for each application audit has a high degree of commonality (except for labelling, instructions for use, or promotional material).
The full application audit assessment fee will apply to one of the applications and a reduced fee will be payable for each of the additional application audits, the degree of which is determined on a case-by-case basis and is at the discretion of the Head, Medical Devices Authorisation Branch.
Assessment fees for mandatory application audits may also be reduced if an IVD (such as an HIV viral load monitoring assay) that is currently registered in the ARTG and therefore has an AUST R is the subject of an application for a Class 3 IVD under the new regulations.
A fee reduction will not apply to transitioning products that are currently listed in the ARTG, i.e. have an AUST L.
A written request for reduced fees and the basis on which abridgement is being sought must be electronically attached to all relevant applications when submitted via eBS.
Regardless of whether a reduction in the assessment fee for an application audit is granted by the TGA, all requests to provide relevant documentation must be met in full and a complete submission package provided. Reference to data provided as part of a previous application for Registration will not be accepted.
Details of the fees currently applicable are available on the TGA website on our Summary of fees and charges to applications submitted to the TGA page.
When an application selected for audit will lapse
In accordance with section 41FK of the Act, an application that has been selected for audit will lapse if:
- the sponsor does not provide the information requested by the TGA within the specified time frame
- the sponsor does not provide a reasonable number of samples of the device, if they have been requested
- the information provided by the sponsor in support of an application is false or misleading
- the sponsor fails to pay the application audit assessment fee.
Information requested for an application audit
The TGA will write to the sponsor requesting submission of the information required to conduct the application audit.
For applications containing multiple IVDs of the same kind that are subject to a mandatory audit, the TGA will generally select one or two of the IVDs from the application to undergo detailed review.
For any additional IVDs that are covered by the application, but which are not selected to undergo a detailed review, the TGA may request limited information, such as a copy of the instructions for use.
Manufacturers may produce multiple IVDs with a high degree of similarity, and therefore it is expected that some will share common technical documentation.
Where appropriate, multiple IVDs may reference a single STED even if they are the subject of different applications for inclusion in the ARTG.
The format and referencing used within a STED covering multiple IVDs must clearly differentiate any similarities and differences.
Documents the sponsor is requested to provide
| Document | Description | Legislative reference/guidance | Note |
|---|---|---|---|
| A copy of the manufacturer's Australian Declaration of Conformity | As part of the conformity assessment procedures, the manufacturer of an IVD is required to make a Declaration of Conformity which declares that the device complies with the Australian legislative requirements. |
| The Declaration of Conformity must be for the Australian requirements. A European declaration of conformity is not acceptable. The Declaration of Conformity may be annexed to the STED |
| Copy of current conformity assessment evidence for the IVD and the manufacturer | Conformity assessment evidence is the certificate(s) issued by a regulatory body, conformity assessment body or certification body that demonstrates:
Conformity assessment evidence is used to demonstrate that an appropriate conformity assessment procedure has been applied by the manufacturer. |
| Includes:
that apply to the class of the IVD. If the manufacturer has applied the conformity assessment procedure for system or procedure packs under Schedule 3, Clause 7.5 of the Regulations, the sponsor may be required to submit copies of the certification for IVDs in the system or procedure pack. Certificates may be annexed to the STED. |
| Summary Technical Documentation (STED) | Manufacturers of all classes of IVDs are expected to demonstrate conformity to the Essential Principles through the preparation and holding of a technical file that shows how each IVD was designed, developed and manufactured. The STED is derived from selected components of the product technical file and provides evidence that the device conforms to the Essential Principles. |
|
|
| The sponsor may also be requested to provide documented evidence for other matters related to the application. |
|
| Example:
|
Components of the STED
Device description
A detailed description of the IVD must be provided, including information addressing each of the following points:
- Intended purpose
- Intended user
- Risk classification according to Australian regulations
- Acceptable specimen types
- Description of principle of the assay and methodology used and
- Description of individual components included in the IVD.
Where applicable, the following should also be provided:
- A description of the specimen collection and/or transport materials required or recommended to be used
- A description of the accessories, other IVDs and other products that are not medical devices which are intended to be used in combination with the IVD
- For assays requiring instrumentation, a description of the relevant instrumentation characteristics or details of dedicated instrumentation to be used
- A description of any software to be used and
- A complete list of any configurations or variants of the IVD, other than kit size, that will be made available.
Where applicable, a review of all platforms/instrumentation and any other materials, including dedicated specimen receptacles, that are required (or recommended) to be used in combination with an IVD will occur in conjunction with this technical file review. Relevant information for every aspect of the IVD should be provided to enable this to occur.
Device history
A summary of the product history in both the Australian market and any other jurisdiction(s) in which it is supplied will be requested to allow the TGA to make an assessment of the safety and efficacy of the IVD in the post-market environment.
Details should include a list of countries or regulatory jurisdictions, approximate numbers of IVDs and/or period of time supplied, summary of any adverse events, recalls, corrective/preventive actions or refusal to approve for supply.
The inclusion of information clearly identifying products either as new to the Australian market, or as previously Registered, Listed or Exempt products transitioning to the requirements of the new IVD regulatory framework will assist the TGA in prioritising the assessment of new products, so as to reduce as much as possible any delay to the market caused by a backlog of IVDs transitioning to the requirements of the new regulations.
Essential Principles checklist
A copy of the Essential Principles checklist that summarises conformity to each applicable Essential Principle by reference to appropriately applied standards, or other appropriate means will be requested.
Evidence of compliance must refer to documents, reports, internal procedures, etc and should include a cross-reference to the location of the documents listed within the checklist.
To establish that an IVD complies with the relevant provisions of the Essential Principles, the TGA may request further information in relation to any of the documents referenced or expected to be held as part of the product technical file.
The TGA will accept a European Essential Requirements checklist to IVDD requirements provided it is also accompanied by a short statement to provide assurance from the manufacturer 'that the Australian Essential Principles, as described in Schedule 1 of the Therapeutic Goods (Medical Devices) Regulations 2002, have been met'.
A template Essential Principles checklist is available from the TGA website.
Risk analysis and control summary
For Class 1-3 IVDs, a summary of the risk management activities performed by the manufacturer of the device must be provided. An example of such a summary is the Risk Management Report required by Clause 8 of ISO14971:2007, however the summary should include as a minimum:
- a list of possible hazards for the IVD arising from false positive or false negative results
- indirect risks which may result from IVD-associated hazards e.g. instability of test components, integrity of packaging, selection of specimens
- the user/operator hazards such as any risks arising from reagents and specimens containing infectious agents and
- the risk mitigation strategies that have been implemented to reduce unacceptable risks.
Taking into account risk mitigating activities, the results of the risk analysis should provide a conclusion that the remaining risks are acceptable when compared to the benefits. The risk analysis and control summary may be submitted either in a summary (text) format or as a reduced table.
Design and manufacturing information
A summary of the design and manufacturing processes at a level of detail appropriate to the risk class of the device should be provided.
The summary should include a review of the design features that make the IVD suitable for its intended purpose, an overview of manufacturing processes and controls, manufacturing sites, a description of critical assay ingredients, a description of the major systems or critical processes, and details of any decision pathways or algorithms used, as appropriate.
Clinical evidence report
Every medical device requires clinical evidence, and for IVDs this represents the information that supports the clinical utility and the performance of the IVD as intended by the manufacturer.
A clinical evidence evaluation report that demonstrates conformity with the applicable provisions of the Essential Principles (as specified in EP14) must be available for all IVDs, other than those that are exempt from inclusion in the ARTG.
The Clinical Evaluation Procedures described in Clause 8, Schedule 3 of the Regulations set out the requirements, and focus on the manufacturer obtaining clinical investigation data through conducting performance evaluations and/or carrying out a literature review of published and unpublished scientific literature.
It is important to note that evidence to support the clinical competence of the author (e.g. short curriculum vitae) must accompany the submitted clinical evidence report to provide assurance that the clinical evidence has been evaluated by a competent clinical expert.
Clinical utility
The clinical utility of a parameter is the demonstration of its potential or established usefulness for patient management decision making, and provides the means for making decisions about effective treatment or preventive strategies.
For many common IVDs with a broad history spanning many years of use, clinical utility has long been established and there are well recognised associations with a particular disease or condition.
For these IVDs it is not expected that extensive information be further documented simply for the purpose of submission for premarket approval.
For more recently developed IVDs which involve the use of a new technology, a new application, a new biomarker, pharmacogenomics, etc, evidence of clinical utility may be required.
Where confirmation of an IVD's clinical utility is required to be documented, the process for generating appropriate evidence should commence at the research phase and often involves ongoing collaborative development over time.
Evidence of clinical utility is initially established using a summary of literature searches and expert opinions, and is supplemented with appropriate clinical or research data as it becomes available.
If a manufacturer considers that evidence of an IVD's clinical utility is not required to be compiled and submitted for review due to its recognised association with a particular disease or condition, this decision is required to be documented and clearly justified as part of the clinical evidence report.
Performance evaluation
Performance evaluation studies incorporate both the clinical and analytical performance characteristics of an IVD. The analytical performance aspects of an IVD's performance evaluation are addressed under the following section - Product validation and verification.
The clinical performance of an IVD is demonstrated by correlating the use of an IVD with a specific clinical condition, in accordance with the target population and intended user.
Clinical performance is a measure of an IVD's ability to correctly identify patients as either having or not having a particular disease or condition, based on their true clinical status.
Clinical performance characteristics include diagnostic sensitivity and diagnostic specificity, which may vary depending on the choice of a cut-off value for the assay, and the negative and positive predictive values which depend on the prevalence of the disease or condition within the population of interest.
For many IVDs, providing data that has been drawn from a clinical performance study or generated within a target population may not always be an essential component of the clinical evidence. For well established and standardised analytes, demonstration of the IVD's analytical performance characteristics may be sufficient to support the use of the IVD as intended by the manufacturer, particularly when the clinical utility for a type of IVD has been long accepted.
Where it is available in a suitable form, published literature or experience gained by routine diagnostic testing which includes post market surveillance data, a summary of adverse events, and details of any field safety corrective actions (recalls, notifications, hazard alerts) may provide sufficient evidence to support the clinical performance of an IVD.
The manufacturer must justify the grounds on which they are circumventing either fully or in part, the requirement to provide clinical performance data wherever this occurs.
For IVDs that are intended to be used by lay persons or at the point of care, it is expected that clinical performance studies take into consideration the level of knowledge, understanding and skills for such users by providing evidence to demonstrate appropriate performance within that target population.
Product validation and verification
Evidence to demonstrate the analytical performance characteristics of the IVD is a requirement under Essential Principle 15 and forms a critical part of the manufacturer's performance evaluation studies, as required for clinical evidence.
The information presented for each study should provide sufficient detail for the assessor to understand how the study was conducted, the characterisation of specimens/samples used, acceptance criteria, explanations for anomalous results, and the outcomes/conclusions drawn.
It is acceptable to combine two or more aspects of analytical performance into fewer separate studies provided each of the studies is well designed and all relevant variables and test characteristics are effectively demonstrated. The following analytical performance characteristics should be specifically addressed, as appropriate to the type of IVD.
Specimen type
A list of all appropriate specimen type(s) suitable for use with the IVD must be provided, including anticoagulants, matrices or any special instructions or conditions associated with specimen collection.
Information should also address specimen stability, appropriate storage conditions and where applicable, transport conditions.
Storage includes elements such as duration, temperature limits, number of freeze/thaw cycles.
Analytical performance study reports should include information about the nature of the specimen types tested (e.g. spiked, wild type etc) and the geographic location where specimens were obtained, as appropriate.
Accuracy
The term accuracy refers to both trueness and precision (reproducibility and repeatability).
Demonstration of trueness requires utilisation of an acceptable reference method or comparison with reference material of a higher order.
Reproducibility should include information about studies to estimate total variability and as appropriate, between-day, between-run, between-sites, between-lots, between-operators and between-instrument variability.
Repeatability should include information about studies to estimate total variability and as appropriate, within-run variability.
The results of testing should include samples that represent the full range of expected analyte concentrations within the target population, and for Class 3 IVDs detailed information is required.
Analytical sensitivity
Demonstration of analytical sensitivity should provide as part of the study design, the analyte tested, how the levels were established, specimen characterisation and number of replicates tested at each concentration. Calculations used to determine the assay sensitivity should be included.
For Class 3 IVDs detailed information is required.
Analytical specificity
Information relating to any studies conducted to determine the effect caused by potentially interfering or cross-reacting substances or agents on test results should be provided.
Consideration should be given to both exogenous and endogenous factors expected to be encountered. For Class 3 IVDs detailed information is required.
Measuring range of assay
A summary of the studies conducted to define the assay measuring range should be included for both linear and non-linear systems. Information provided should describe the lower limit of detection and how this was determined (e.g. preparation of dilutions, standards, number of replicates) and include an investigation into any potential effects of prozone or high-dose hook effect, if applicable.
Traceability of calibrator and controls
Information summarising the traceability of calibrators and trueness control materials should be provided, if applicable. Methods used to determine traceability to reference material of a higher order, acceptance criteria, and the assignment and validation of values should be included.
Determination of assay cut-off
Where applicable, a summary of the process used to establish the assay cut-off should be provided. Information provided should be based on the population studied, method(s) used to establish the true status and any statistical methods used to generate results e.g. Receiver Operator characteristic (ROC) curve.
Verification and validation of instrumentation/software
For verification and validation of instrumentation and/or software IVDs, the study report should include a summary of performance testing undertaken conducted in a valid end-user environment.
Stability
Stability studies to support the claimed shelf-life under closed, in-use and transport conditions must be provided.
For Class 3 IVDs a copy of the study protocol, a detailed study report showing the results of testing, any calculations performed and conclusions drawn should be included.
For Class 2 IVDs a summary report detailing the nature of the stability study conducted, any anomalous results/investigations, and a conclusion which supports the proposed shelf life and storage conditions is considered acceptable.
For closed shelf-life studies, data must be generated by testing at appropriate storage time intervals using a minimum of 3 separate production batches of IVDs.
Temperature ranges assigned for testing should encompass both the upper and lower storage temperatures claimed. Real time data which extends beyond the proposed shelf life should be provided for at least 1 batch of product.
Accelerated data generated using product stored under exaggerated conditions (including elevated temperature, high humidity, increased light and vibration, as appropriate) will be accepted for subsequent batches of product as an interim measure until such time as real time studies can be completed.
Ongoing real time studies for those products where the shelf-life has been assigned on the basis of partially accelerated data should be monitored closely, and the manufacturer should reduce the shelf-life in line with the real-time data as appropriate.
For newly released products only, where insufficient time has passed to allow at least one production batch of product to undergo appropriate real time stability testing since the time of release, the TGA may give consideration to the assignment of a nominal (reduced) shelf life on the basis of accelerated stability studies, provided they have been conducted under exaggerated conditions that include elevated temperature, high humidity, increased light and vibration, as appropriate.
Ongoing real time studies are required to be monitored closely.
In-use (open vial) stability and transport simulation studies should be conducted using at least one batch of product, with a study design which includes conditions appropriate to the intended use and expected conditions likely to be encountered for the product.
For further information refer to CLSI standard EP25-A, Evaluation of Stability of In Vitro Diagnostic Reagents.
Information to be supplied with the IVD
The sponsor is required to provide clear, legible copies of representative information that is to accompany the kind of IVD medical device when supplied in Australia, including:
- Labelling
- Instructions For Use and
- Advertising material (e.g. brochures, webpages, published advertisements, etc.), where available.
Labelling and instructions for use are not necessarily required for every model or variation, unless there are significant differences in content. However, the copies provided are required to be representative of what will be supplied in Australia.
The sponsor's name and address must be provided with the IVD in such a way that the user can readily identify the sponsor. Labelling requirements are prescribed in Regulation 10.2 and Essential Principle 13.2 in Schedule 1.
All representative information must be provided in English.
Depth of information to be provided
The following table summarises the depth of detail required to be contained in the STED.
References to Class 4 IVDs in this table indicate the level of detail expected in the STED for products undergoing a design examination.
Class 4 IVDs must be covered by a TGA Conformity Assessment Certificate and are not required to undergo application audit.
| Section | Class 1 | Class 2 | Class 3 | Class 4 |
|---|---|---|---|---|
| Device description including variants | ||||
| Device description | Address each point - all classes | |||
| Reference to previous device generation - not yet available on any market | Summary | Summary | Summary | Summary |
| Device history - already available on the market in another jurisdiction | Summary | Summary | Summary | Summary |
| Risk analysis and control | Summary or Reduced table | Detailed | ||
| Design and manufacturing information | ||||
| Device design | Summary | Summary | Summary | Detailed |
| Manufacturing processes | - | - | - | Summary |
| Design and manufacturing sites | Summary | Summary | Summary | Summary |
| Product validation and verification | ||||
| Specimen type | Summary | Summary | Summary | Detailed |
| Accuracy - Trueness | Summary | Summary | Detailed | Detailed |
| Precision - Reproducibility and Repeatability | Summary | Summary | Detailed | Detailed |
| Traceability of control and control materials | Summary | Summary | Summary | Detailed |
| Analytical sensitivity | Summary | Summary | Detailed | Detailed |
| Analytical specificity | Summary | Summary | Detailed | Detailed |
| Measuring range of the assay | Summary | Summary | Detailed | Detailed |
| Validation of assay cut-off | Summary | Summary | Detailed | Detailed |
| Stability | ||||
| Claimed shelf-life | Summary | Summary | Detailed | Detailed |
| In use stability | Summary | Summary | Detailed | Detailed |
| Shipping stability | Summary | Summary | Detailed | Detailed |
| Software | Summary | Summary | Summary | Detailed |
| Clinical evidence | Summary | Summary | Detailed | Elaborated |
The following information provides explanations for the terms used in the table to describe the depth of detail required in the STED:
Summary information
- Brief description of protocol
- Study results
- Study Conclusion
Detailed information
- Study protocol
- Method of data analysis
- Study report (summary of external reports)
- Study Conclusion
Elaborated information
- Study protocol
- Method of data analysis
- Study report (all external reports)
- Study Conclusion
- Raw/line data
Further information may also be found in the GHTF document: Summary Technical Documentation (STED) for Demonstrating Conformity to the Essential Principles of Safety and Performance of In Vitro Diagnostic Medical Devices
The following table shows the terms used for the various devices classes in the GHTF document and their equivalents for IVDs under the Australian regulations.
| GHTF IVD Class | Australian IVD Class |
|---|---|
| Class A | Class 1 |
| Class B | Class 2 |
| Class C | Class 3 |
| Class D | Class 4 |
General requirements for the information to be supplied
We require all the requested information to be provided as a complete stand-alone submission.
Cross-referencing to information submitted in support of previous applications that are already included in the ARTG or are still being processed is not acceptable.
Two hard copies of the documentation are normally requested.
An additional copy in electronic format (on CD or DVD) is highly desirable and may assist us with the assessment.
The sponsor should ensure that:
- a copy of the eBS application form is included
- the supporting information is supplied in loose-leaf binders - plastic sleeves or stapled material should not be submitted
- the information is sectioned for ease of reference, and a table of contents provided which details the content of the binder(s)
- there are appropriately named tab identifiers. For example, the Labelling information should be separated from the other documents by a tab identifier named Labelling
- each page is sequentially numbered
- standard A4 paper is used for all submissions wherever practicable. Text and tables should be prepared using margins that allow the document to be printed on A4 paper, or A3 paper where a larger format is necessary. The left hand margin should be sufficiently large that information is not obscured through binding.
- font sizes for text and tables are of a style and size that are large enough to be easily legible, even after photocopying or when provided electronically. Fonts smaller than 12 points should be avoided whenever possible, except in tables and footnotes where a font size of 10 points is acceptable.
- information supporting an application is in English and legible. Where material is not originally in English a full translation must be submitted, the accuracy of which is the responsibility of the sponsor.
- Australian legal units of measure (SI units) are used. Units generally accepted in clinical practice may also be used (e.g. IU/mL).
- all text and drawings are legible and drawings are clearly labelled.
The applicant should ensure that they comply with the request for information that is issued under s41JA. The formats suggested above are indicative only and may change from time to time.
Timeframe for the provision of information
The Act and Regulations require that the sponsor either hold documentation to substantiate compliance with the Essential Principles or have in place procedures to obtain that documentation from the manufacturer within 20 workdays.
The sponsor is required to certify that they have procedures in place to address these requirements when they submit the application to include an IVD in the ARTG.
It is suggested that before applying for an IVD which is expected to undergo a mandatory application audit, a sponsor may benefit by seeking from their manufacturer in advance the necessary documentation outlined in this guidance document.
This will ensure that they are ready to proceed once a request from the TGA to provide further information is received and allows time to deal with any contingencies that were not anticipated.
When the TGA writes to the sponsor to request the documentation, the letter will specify a date that the information is required by.
If the requested documentation:
- is not received by the specified date; or
- does not meet the requirements outlined in the letter;
then the application will lapse.
To supply the device in Australia the sponsor will then need to re-apply to include the device in the ARTG and pay the associated fees again.
Where to send the information
By Post
Administration Officer
IVD Assessment Section
Medical Devices Authorisation Branch
Therapeutic Goods Administration
PO Box 100
WODEN ACT 2606 AUSTRALIA
By Courier
Coordination Team
IVD Assessment Section
Medical Devices Authorisation Branch
1 Tindal Lane
CANBERRA AIRPORT ACT 2609 AUSTRALIA
By Email
For less than 10 MB in size email files as an attachment to: IVDs@tga.gov.au.
For submissions larger than 10 MB in size email eSubmissions@health.gov.au and provide contact details.
On receipt of these details, we shall contact you to arrange registration for our temporary electronic upload facility.
We also accept electronic data in the form of a USB - delivered to the address provided in your request letter.
We do not accept hard copy data.
Footnotes
- In the context of IVDs, the terms "application audit" and "technical file review (TFR)" are often used interchangeably. It should be noted however that an application audit may be more accurately described as a review of both the administrative and the technical aspects of an application, to establish that the IVD complies with the legislated requirements and that the matters certified by the applicant are correct. Conversely, a TFR refers principally to the examination of the technical documentation which has been prepared to demonstrate compliance with the Essential Principles.
Page history
Title changed from 'Application audit (technical file review) of IVD medical device applications' to 'Preparing for technical file review (application audit) for in-vitro diagnostic (IVD) medical devices' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updated contact details.
Original publication.
Title changed from 'Application audit (technical file review) of IVD medical device applications' to 'Preparing for technical file review (application audit) for in-vitro diagnostic (IVD) medical devices' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updated contact details.
Original publication.