Releasing medicines manufactured at multiple sites: good manufacturing practice (GMP)
Guidance for interpreting the Pharmaceutical Inspection Co-operation Scheme (PIC/S) guide to good manufacturing practice (GMP) for medicinal products.
Purpose
If you are a sponsor or manufacturer of a medicine manufactured across multiple sites, then this guidance will help you understand:
- release for further processing (RFFP) requirements
- how RFFP fits into the release for supply (RFS) process.
Read this guidance together with the general requirements and responsibilities of release for supply of medicines, which apply to Australian sponsors and all TGA licensed or certified manufacturers.
Scientific guideline
Release for further processing (RFFP)
RFFP refers to release of either:
- an intermediate or bulk product to the next manufacturer in the supply chain
- the finished product that requires alteration for distribution into the Australian Market.
Such arrangements are to be defined in the 'GMP agreements' section of this guidance.
Development of this guidance
We have developed this guidance to demonstrate how the release for supply process works across multiple sites of manufacture:
- in response to requests from industry for further guidance
- in consultation with experts from the relevant industry associations.
Scenarios to demonstrate RFFP and RFS
This guidance provides scenarios to help demonstrate some of the issues for consideration in order to meet the general RFS requirements, particularly for:
- specific areas of manufacture, for example listed and complementary medicines
- complex supply chains where manufacture occurs at multiple sites
Updating scenarios
This guidance is a living document. We can update it with scenarios that are more specific, where further interpretation is required. Any updates will only occur after appropriate consultation with the relevant stakeholders.
Intention
This information is provided for guidance only and has been developed on the basis of current knowledge of the subject matter. It should not be relied upon to address every aspect of the relevant legislation. Please also refer to the Therapeutic Goods Act 1989 and the Therapeutic Goods Regulations 1990 for legislative requirements and the current version of the PIC/S Guide to GMP.
Content of this guidance
This guidance covers:
- how to meet RFS requirements when manufacturing at multiple sites, including:
- Sponsor responsibilities
- Sponsor and manufacturer – joint responsibility
- Manufacturer responsibilities
- RFS authorised person responsibilities
- RFFP authorised person responsibilities
- GMP agreements
- RFFP documentation requirements
- minimum RFFP documentation package
- Certificate of Analysis
- scenarios to demonstrate release for supply when manufacturing at multiple sites:
- Scenario 1 - RFS from secondary packaging site
- Scenario 2 - Two consecutive RFS steps.
For information on release for supply of medicinal gases, refer to the medicinal gas guidance.
Related information
Related information:
- PIC/S Guide to GMP Part II: RFFP of active pharmaceutical ingredients and intermediates Section 6.7 Batch production record review.
GMP requirements when manufacturing at multiple sites
The information presented in this section aims to help you understand how to apply the RFS requirements when complex manufacturing occurs, such as manufacturing at multiple sites and how release for further processing fits into the RFS process.
Sponsor responsibilities
The responsibilities listed here are in addition to all of the sponsor responsibilities, and joint responsibilities with the manufacturer, outlined in the release for supply of medicines guidance.
The sponsor is responsible for ensuring that:
- each ARTG entry has at least one TGA approved manufacturer (i.e. licensed or holding a current GMP clearance) responsible for each manufacturing step, including the RFS manufacturing step.
Traceability of release for supply
Where the ARTG entry of a medicine allows more than one RFS site of the finished product batch, the Australian sponsor is responsible for ensuring:
- the ability to identify:
- the site at which any particular batch has been released for supply
- the name of the authorised person responsible for releasing each batch
- inclusion in the batch records (for review by the authorised person conducting RFS) both the RFS site and RFS authorised person
Statements related to the shelf life and stability data
Sponsors are responsible for ensuring the authorised person performing RFS receives signed and dated written statements regarding the shelf life and stability for:
- Finished product:
- a statement that summarises current approved shelf life allocated to the market authorisation and the current status of the ongoing stability testing for the finished product OR
- a statement that summarises initial stability data of the finished product used to define the shelf life of the batch of finished product OR
- a statement that justifies the applied shelf life of the finished product when initial long-term stability testing of listed medicines is being undertaken
- Intermediate or bulk product stored for more than three months:
- a statement that summarises the initial stability data of the intermediate or bulk product used to define the shelf life and the current status of the ongoing stability testing of the intermediate or bulk product OR
- a statement that justifies the applied shelf life of the intermediate or bulk product when undertaking initial long term stability testing of the intermediate or bulk product
Sponsor and manufacturer - joint RFFP responsibility
The PIC/S Guide to GMP identifies shared responsibilities between the sponsor (marketing authorisation holder) and the manufacturer.
Provide RFFP authorised person access to information
Where manufacture occurs across multiple sites the authorised person, performing RFFP to the next manufacturer in the supply chain, requires access to all information that is relevant to the manufacturing steps for which they are responsible.
Provide the authorised person(s) performing RFFP full access to all relevant aspects of:
- the marketing authorisation, including details in the ARTG and other matters agreed on in writing between TGA and the Australian sponsor
- the manufacturing steps, if a TGA-licenced manufacturer, including:
- the licence to manufacture therapeutic goods
- all authorisations and conditions under the licence
- the steps in manufacture granted under section 38, Therapeutic Goods Act 1989 and conditions of licences as imposed under section 40, Therapeutic Goods Act 1989
- the manufacturing steps, if an overseas manufacturer:
- the TGA GMP certificate and GMP clearances
- all authorisations and conditions, as imposed under sections 25(1)(g), 26(1)(g) and 26A(3, Therapeutic Goods Act 1989
- the PIC/S Guide to GMP as specified in the current manufacturing principles
- default standards under section 10 of the Therapeutic Goods Act 1989
- all applicable Therapeutic Goods Orders
Manufacturer responsibilities
In addition to the manufacturer RFS responsibilities, for medicines manufactured at more than one site, each manufacturer in the supply chain is to ensure their PQS contains:
- detailed procedures and responsibilities for RFFP to the next manufacturer in the supply chain, where applicable
- detailed release for supply procedures, where applicable
RFS authorised person responsibilities
Release for supply of medicines outlines the responsibilities of an authorised person performing RFS. When manufacture occurs at multiple sites, the authorised person performing RFS of the finished product batch is allowed to rely on decisions of delegated authorised persons.
Delegated authorised persons
A delegated authorised person can either be working:
- within the same company (on the same site or on a different site) OR
- with another manufacturer
A delegated authorised person has specific responsibilities when performing RFFP.
Additional responsibilities of RFS authorised persons
When manufacture occurs at multiple sites, there are additional responsibilities for the authorised persons performing RFS, including:
- reviewing the complete RFFP documentation
- relying on decision of other authorised persons.
Relying on decisions of other authorised person(s)
The authorised person performing release for supply can only rely on decisions by other authorised persons when all necessary steps have been completed through an appropriate PQS and all of the following conditions are met:
- All authorised person(s) have full access to all parts of the marketing authorisation that are relevant for the manufacturing steps performed at the site for which they are the authorised person.
- All partial manufacturers must be covered by valid GMP agreements that define RFFP to the next manufacturer in the supply chain.
- The PQS at each manufacturing site has detailed information on the release procedures.
- All decisions on RFFP to the next manufacturer in the supply chain are recorded through a legally valid signature. For example, a full written signature either:
- on a paper document OR
- an electronic signature in a validated electronic environment (note Annex 11 of the PIC/S Guide to GMP)
- The authorised person performing RFS has accepted the PQS used for RFFP, for example through the supplier qualification program.
- The authorised person at each site performing RFFP is required to fulfil the RFFP authorised person responsibilities described below.
The level of evidence required for the authorised person responsible for RFS to be confident of compliance with these conditions depends on the type of products manufactured.
RFFP authorised person responsibilities
All RFFP decisions are made by an authorised person, or delegate, including a review of the supporting documentation package.
The responsibilities of the RFFP authorised person are to:
- have access to the all parts of the marketing authorisation that are relevant for the steps in manufacture performed at the site for which they are the authorised person
- follow the release procedures described in the PQS
- record all decisions regarding RFFP through a legally valid signature, for example:
- full written signature on a paper document
- electronic signature in a validated electronic environment (note Annex 11 of the PIC/S Guide to GMP)
- complete the RFFP documentation requirements
- ensure the RFS authorised person receives all RFFP documentation prior to RFS by:
- sending all RFFP documentation for each batch, including RFFP documents from previous manufacturing steps, to the authorised person at the next manufacturing site
- providing all relevant product quality review (PQR) data to the manufacturer performing RFS
Where two consecutive RFS steps occur, refer to the 'scenario 2' section of this guidance.
GMP agreements
Sponsors and manufacturers are required to establish valid GMP agreements to cover all partial manufacturers in the supply chain, in accordance with the PIC/S Guide to GMP.
Valid GMP agreements
Valid GMP agreements are to:
- clearly define the RFFP to the next manufacturer in the supply chain, as applicable
- clearly establish responsibilities for all GMP related activities at each licenced or certified site.
- specify how the RFS authorised person ensures that each batch has been manufactured and checked for compliance with the marketing authorisation, where applicable
- include an obligation on all providers of bulk or intermediate product to notify the recipient(s) of any:
- significant deviations from the agreed production process
- out-of-specification results
- non-compliance with GMP
- investigations
- complaints
- other matters that should be taken into account by the RFS authorised person.
Ongoing stability programs
The manufacturer(s) performing release for supply and the sponsor share the responsibility to ensure an ongoing stability program of the finished product is conducted.
GMP agreements are to detail these mutual responsibilities, by ensuring:
- a mechanism is described for how the authorised person conducting RFS obtains access to the ongoing stability data as required, e.g. inspections, recalls, complaints
- a mechanism is described for how the TGA inspector obtains access to the ongoing stability data during an inspection of the RFS manufacturer, if the RFS manufacturer is not the compiler of stability data
Contracted services
You can contract out ongoing stability studies, but both the RFS manufacturer and the sponsor need to be able to access and review the results.
Product quality reviews (PQR)
GMP agreements are to detail the mutual responsibilities concerning the PQR, ensuring:
- which manufacturer in the supply chain, or the sponsor, is responsible for compiling the PQR data into the complete PQR
- the process for communicating all the relevant PQR information to the compiler
- a mechanism to ensure that the full PQR is accessible by the authorised person at the site undertaking RFS, as required, e.g. inspections, recalls, complaints
- a mechanism for the TGA inspector to obtain the PQR during an inspection of the RFS manufacturer, if the RFS manufacturer is not the PQR compiler.
Arrangements for collecting PQR data
Where multiple manufacturers are involved in the manufacture of a product:
- each (contract) manufacturer will normally:
- prepare the data for the PQRs of the manufacturing steps performed by that manufacturer
- submit the relevant PQR data to the manufacturer performing RFS of the finished product batch
- the manufacturer responsible for RFS is responsible for:
- collating the data into complete PQRs
- the authorised person performing RFS must consider the results within the context of the PQR findings
You can consider other arrangements, as long as all the relevant data are provided to one manufacturer in the supply chain or to the sponsor who prepares complete PQRs. In that case, you must clearly define the arrangements in all applicable GMP agreements.
RFFP documentation requirements
This information is relevant when manufacture of a batch of product occurs at more than one manufacturing site.
The authorised person at each manufacturing site is required to:
- complete an identical minimum RFFP documentation package
- send the RFFP documentation package to the next authorised person in the chain of manufacture
- accumulate all of the RFFP documents so that the authorised person performing RFS can review the full set of RFFP documents
Minimum RFFP documentation package
The minimum RFFP documentation package is to be printed showing the company letterhead and signed and dated by the authorised person. If you are using computer systems to record RFFP decisions, take note of Annex 11 of the PIC/S Guide to GMP.
The RFFP documentation package comprises:
- product name
- product code
- batch number
- assurances that:
- the step(s) in manufacture for the batch have been undertaken in compliance with Australian marketing authorisation, where applicable
- any deviations that may impact medicine quality, efficacy or shelf life have been resolved
- there are no investigations, complaints or other matters that may impact product quality, efficacy or shelf life that should be taken into account by the authorised person responsible for RFS
- the storage conditions on the label are consistent with the shelf life of the finished product
- quality risk management principles were applied to develop an approach tailored to the risk situation
- the information and data relating to manufacture of the batch has been added to the PQR program at the site and that an annual review of that information and data is part of that program
- Acknowledgement that a mechanism has been established to allow relevant PQR and ongoing stability data and reviews to be provided to the authorised person at the site performing the RFS step in manufacture.
The sponsor is responsible for ensuring that the authorised person conducting RFS receives appropriate information about shelf life and stability data.
Depending on the circumstance, stability data may not always be required, such as described in the 'Scenario 1 – RFS from a secondary packaging site' section of this guidance, where re-release for supply occurs after minor further steps at a second site.
Certificate of Analysis
The entity with GMP responsibility for compiling the testing results of the batch is also responsible for adding the relevant Certificate of Analysis for each batch to the accumulating RFFP documentation package.
Release for supply scenarios when manufacturing at multiple sites
These scenarios relate to medicines manufactured at multiple manufacturing sites.
The scenarios provided:
- aim to demonstrate how RFFP and RFS steps relate to each other
- do not represent all possible scenarios
- are designed to be updated, where additional information is requested from industry.
The addition of specific examples of different scenarios are welcome. To request inclusion:
- discuss with your Industry Association in the first instance
- the Industry Association will then discuss the request with us.
Updates or additional scenarios will only occur in consultation with relevant stakeholders.
We describe the following scenarios:
Scenario 1 – RFS from a secondary packaging site
In this scenario, release for supply occurs at a secondary packaging site (Site 2). The primary packaging occurs at a different site (Site 1) and is released for further processing.
Scenario 2 – Two consecutive RFS steps
In this scenario, release for supply occurs twice. After RFS, the product requires relabelling or additional secondary packaging, requiring a second RFS step.
We provided an additional scenario for medicinal gases in our medicinal gas guidance.
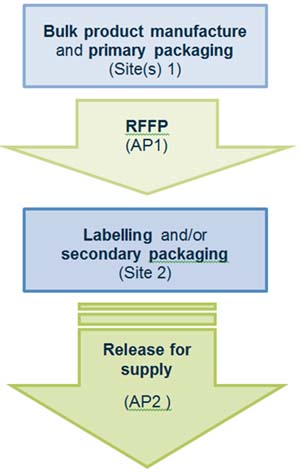
Scenario 1 - RFS from a secondary packaging site
In this scenario:
Manufacturing steps that occur at site(s) 1 include:
- bulk product manufacture and primary packaging
- release for further processing, performed by authorised person 1 (AP1)
Manufacturing steps that occur at site 2 include:
- labelling and/or secondary packaging
- release for supply of the finished product, performed by authorised person 2 (AP2)
Specific examples of labelling or secondary packaging steps include:
- a bottle is placed in a carton
- the Consumer Medicine Information (CMI) is added to the product
- the medicine is over-labelled or relabelled
- a carton is added or replaced
- the pack size is changed without changing the primary pack, e.g. changing the number of blister cards in a pack
Flowchart for release for supply from a secondary packaging site.
{kind=link}
Flow chart for release for supply from a secondary packaging site:
- Bulk product manufacture and primary packaging occurs at site(s) 1.
- Release for further processing is performed by authorised person 1 at site(s) 1.
- Labelling and/or secondary packaging occurs at site 2.
- Release for supply by authorised person 2 occurs at site(s) 2.
Description of specific issues
In this scenario:
- The labelling and/or secondary packaging performed at the secondary site did not alter the primary packaging or the stability profile.
- Additional stability would not normally be required, as the stability profile is not changed.
- Site 1 would be required to take responsibility for:
- ongoing stability for the primary packaged product
- compiling the full PQR.
- Authorised person 1 at site 1 performing RFFP has:
- certified that the bulk and primary packaging was manufactured in accordance with the market authorisation, where applicable
- provided authorised person 2, conducting RFS at site 2, with the complete RFFP documentation.
Meeting GMP compliance
RFFP authorised person
The authorised person performing RFFP is to perform all duties of an authorised person as outlined in RFFP authorised person responsibilities.
In this scenario, authorised person 1, performing RFFP, is required to provide the minimum RFFP documentation to the RFS authorised person 2, including:
- a Certificate of Analysis for the batch
- a batch specific certification for compliance to the Australian marketing authorisation for the steps of manufacture covered. This should include, for example but not limited to:
- assurance that the batch has been manufactured in accordance with the marketing authorisation
- assurance that any deviations that may impact medicine quality, efficacy, safety and shelf life have been resolved
- verification that product storage has met requirements.
- a declaration that the PQR and ongoing stability data have been performed, are current, and meet specifications, GMP requirements and the marketing authorisation
RFS authorised person
The authorised person performing RFS at Site 2 is to performing all duties of an authorised person for RFS as outlined in release for supply of medicinal products
In this scenario, authorised person 2 performing RFS is required to rely on the RFFP documentation package from authorised person 1 at site(s) 1.
Risk assessment
In this scenario, we expect a formal risk assessment to be completed, prior to the secondary packaging steps, to demonstrate that the:
- additional steps represent no further risk to the product
- modifications being performed either:
- have no impact on stability
- has an impact on stability, in which case, authorised person 2 is expected to verify the suitability of the secondary packaging process
Ongoing stability
The manufacturer performing release for supply and the sponsor need to fulfil the requirements detailed in their GMP agreement.
In this scenario:
- all applicable aspects of manufacture, up to the step of RFFP from site(s) 1, were completed in accordance with PIC/S Guide to GMP requirements
- the additional steps at Site 2:
- represent no further risk to the product
- do not change the product expiry date
In this scenario, staff at the second RFS site (Site 2) do not need to hold ongoing stability data, if all risks and conditions have been covered, as described in this scenario.
Product quality review
The PQR is required to cover all relevant steps of manufacture, including the packaging steps. The manufacturer performing RFS and the sponsor need to fulfil the requirements detailed in their GMP agreement.
In this scenario, the established GMP agreement could specify that:
- authorised person 1 at site 1 is responsible for compiling the full PQR
- authorised person 2 at site 2 communicates any information from the secondary packaging step, relevant to compiling the PQR, to authorised person 1 at site 1
- full PQR information will be accessible to authorised person 2 performing RFS at site 2, as required e.g. inspection, recalls, complaints etc.
The site undertaking RFS (Site 2) does not need to hold the full PQR information if:
- all risks and conditions have been addressed and documented as outlined in this scenario
- the GMP agreement ensures the full PQR is accessible by the authorised person at the site undertaking RFS, as required, e.g. inspections, recalls, complaints.
Information for TGA inspectors
In this scenario, during an inspection of Site 2, the TGA inspector will need access to ongoing stability data and PQRs.
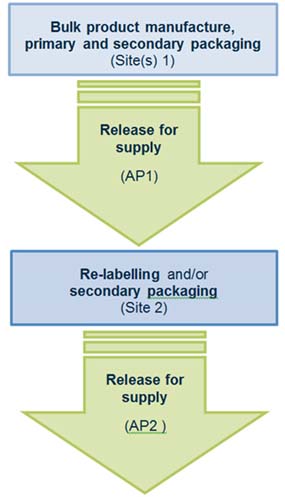
Scenario 2 – Two consecutive RFS steps
In this scenario, RFS occurs twice. Following the first RFS, the product undergoes relabelling or additional secondary packaging steps. These extra steps require an additional RFS step.
The following manufacturing steps occur at site(s) 1:
- the bulk product manufacture, primary and secondary packaging
- release for supply is conducted by authorised person 1 (AP1).
The following manufacturing steps occur at site 2:
- re-labelling and/or secondary packaging
- release for supply conducted by authorised person 2 (AP2).
A secondary RFS step due to relabelling or secondary repackaging may occur, in examples such as but not limited to, when:
- the product is modified following a change to marketing authorisation, requiring:
- addition of changed CMI
- over-labelling or relabelling
- carton change or add an additional carton
- change or add an additional carton.
Flowchart for two consecutive release for supply steps
{kind=link}
- Bulk product manufacture, primary and secondary packaging occurs at site(s) 1.
- Release for supply conducted by authorised person 1, at site(s) 1.
- Re-labelling and/or secondary packaging occurs at site 2.
- Release for supply conducted by authorised person 2, at site 2.
Description of specific issues
In this scenario:
- the primary packaging is not altered between the two RFS steps, so there is no change to the stability profile
- no changes are made to the product expiry date between the first RFS step and the second RFS step
- at the first RFS step, the authorised person certified that manufacturing of the product occurred in accordance with both:
- the marketing authorisation
- the PIC/S Guide to GMP requirements.
In this situation, it is acceptable that the authorised person 2, performing the RFS from site 2, relies on the certification and supporting documentation from the first RFS step from site 1.
Addressing GMP compliance
First RFS
The authorised person performing RFS is to performing all duties of an authorised person for RFS as outlined in release for supply of medicinal products
In this scenario, authorised person 1, conducting RFS from site 1, provides the RFS certification to the authorised person at site 2, as the product had already been released.
Second RFS
The authorised person 2, performing RFS from site 2:
- is also required to perform all the duties of an authorised person outlined in release for supply of medicinal products
- requires receipt of the initial RFS certification from site 1
- is required to address the issues below
Risk assessment
In this scenario, a formal risk assessment is required, prior to the repackaging steps, clearly demonstrating that the:
- modifications being performed have either:
- no impact on stability
- an impact on stability and the suitability of the secondary packaging process is verified by authorised person 2
- information generated is suitable for:
- verifying the consistency of the existing process
- verifying the appropriateness of current specifications for both starting materials and finished product
- highlighting any trends
- identifying possible product and process improvements
The additional steps should create no extra risk.
Ongoing stability data
The manufacturer performing release for supply and the sponsor need to fulfil the requirements detailed in their GMP agreement.
In this scenario, the established GMP agreement could specify that ongoing stability data will be accessible to authorised person 2 at the site conducting secondary re-packaging and undertaking release for supply, as required e.g. inspection, recalls, complaints etc.
In this scenario, staff at the second RFS site (Site 2) do not need to hold ongoing stability data, if all risks and conditions have been covered, as described in this scenario.
Product quality review
The PQR is required to cover all relevant steps of manufacture, including the packaging steps. The manufacturer performing RFS and the sponsor need to fulfil the requirements detailed in their GMP agreement.
In this scenario, the established GMP agreement could specify that:
- authorised person 1 is responsible for compiling the full PQR
- the secondary re-packaging manufacturer prepares the PQR for their step of manufacture and provides this to authorised person 1 to prepare the full PQR
The site undertaking RFS (Site 2) does not need to hold the full PQR information if:
- all risks and conditions have been addressed and documented, as outlined in this scenario
- the GMP agreement ensures the full PQR is accessible to the authorised person at the site conducting secondary re-packaging and undertaking RFS, as required, e.g. inspections, recalls, complaints
Information for TGA inspectors
During an inspection of Site 1 or Site 2, the TGA inspector needs access to ongoing stability data and PQRs.
Page history
Title changed from 'Releasing medicines manufactured at multiple sites' to 'Releasing medicines manufactured at multiple sites: Good Manufacturing Practice (GMP)' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updated to link to PIC/S Guide to GMP for medicinal products – version 16.
Removal of final bullet point (and related sentences) under Scenario 2 – page 18.
- Restructured with new title
- Updated to be consistent with PE009-13
Guidance on release for supply for medicinal product manufacturers Part 2.
Title changed from 'Releasing medicines manufactured at multiple sites' to 'Releasing medicines manufactured at multiple sites: Good Manufacturing Practice (GMP)' as part of migration to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Updated to link to PIC/S Guide to GMP for medicinal products – version 16.
Removal of final bullet point (and related sentences) under Scenario 2 – page 18.
- Restructured with new title
- Updated to be consistent with PE009-13
Guidance on release for supply for medicinal product manufacturers Part 2.