Australian clinical trial handbook
Guidance on conducting clinical trials in Australia using 'unapproved' therapeutic goods.
We are currently updating and restructuring our clinical trial webpages. This page will be updated as improvements are made.
Purpose
This handbook provides guidance on the legislative, regulatory and good clinical practice (GCP) requirements when conducting clinical trials in Australia using 'unapproved' therapeutic goods. It assists trial sponsors, Human Research Ethics Committees (HRECs), investigators and approving authorities (institutions) to understand their roles and responsibilities under the therapeutic goods legislation.
Information about clinical trials for consumers can be found on the Australian Clinical Trials website.
This handbook does not describe all of the requirements for conducting clinical trials in Australia. It refers to other relevant publications throughout that should be read in conjunction with this guidance.
This handbook describes the two schemes under which clinical trials involving 'unapproved' therapeutic goods may be conducted in Australia:
- Clinical Trial Notification (CTN) scheme
- Clinical Trial Approval (CTA) scheme.
Clinical trials that do not involve the use of 'unapproved' therapeutic goods are not subject to the requirements of the CTN and CTA schemes.
This guidance has been developed by the Therapeutic Goods Administration (TGA) and therefore the use of ‘we’ and ‘us’ throughout refers to TGA.
See the TGA glossary for definitions relevant to the regulation of therapeutic goods in Australia.
Legislation
Clinical trials involving therapeutic goods
Clinical trials involving therapeutic goods are generally undertaken to assess the effects, efficacy, performance and/or safety of the product. It is therefore necessary that clinical trials are conducted using appropriate experimental designs to obtain valid data without exposing participants to unnecessary risks.
Before starting a clinical trial, all parties should be satisfied that the rights, safety and well-being of trial participants will be protected and that clinical trial data generated will be reliable and robust. To achieve such objectives, clinical trials involving ‘unapproved’ therapeutic goods must be conducted in accordance with:
- the requirements of the therapeutic goods legislation
- the principles that have their origin in the World Medical Association Declaration of Helsinki
- the National Statement on Ethical Conduct in Human Research (the National Statement) as in force from time to time
- the relevant Good Clinical Practice (GCP) guideline
- other relevant requirements of Commonwealth and/or state and territory legislation
- site specific requirements.
Before conducting a clinical trial in Australia, the trial sponsor will need to consult a HREC to determine whether an exemption under the CTN scheme or approval under the CTA scheme is required for the trial.
Clinical trials that do not involve the use of ‘unapproved’ therapeutic goods are not subject to CTN or CTA requirements. However, all clinical trials require HREC approval before the clinical trial may commence.
The trial sponsor will need to determine the following for each product used in the clinical trial:
- is the product a therapeutic good (and therefore regulated as a therapeutic good)
- the type of therapeutic good (and therefore regulated as a medicine, medical device or biological)
- If the product is considered ‘unapproved’ (and therefore a CTN or CTA submission is required).
Is the product a therapeutic good?
The trial sponsor must first determine whether any of the products to be used in a clinical trial meet the definition of a therapeutic good and are regulated under the therapeutic goods legislation.
The following guidance may assist trial sponsors to determine if a product is a therapeutic good:
- What are ‘therapeutic goods’? provides definitions and examples of therapeutic goods
- Is my product a therapeutic good? is an online tool designed to assist in determining whether a product is a therapeutic good, and if so, the type of therapeutic good (although this is not specifically designed for clinical trials)
- Food and medicine regulation and the Food-Medicine Interface Guidance Tool may assist in identifying whether a product is regulated as a medicine or a food.
Section 3 of the Therapeutic Goods Act 1989 provides definitions for therapeutic goods and therapeutic use. Although the term ‘therapeutic use’ is sometimes used in connection with phase IV clinical trials for medicines and biologicals, for the purposes of the CTN and CTA schemes, the use of a medicine or biological in a phase I, II or III clinical trial will also generally be considered to be for therapeutic use. A medicine, biological or medical device that is the subject of a clinical trial will generally be considered to be a therapeutic good regardless of the phase or stage of the trial.
Determine the type of therapeutic good
The trial sponsor needs to correctly identify the type(s) of therapeutic good(s) being supplied in a clinical trial as different regulatory requirements apply.
This handbook describes the regulation of three main types of therapeutic goods:
- medicines (including prescription medicines, over-the-counter medicines and complementary medicines)
- medical devices (including in vitro diagnostic medical devices (IVDs))
- biologicals (including human cell and tissue-based therapeutic goods, or live animal cells, tissues and organs).
See What is regulated as a biological for information on therapeutic goods that are regulated as biologicals. Biological prescription medicines (for example, vaccines that do not contain human cells, plasma derivatives, recombinant products) are not regulated by us as biologicals. They are regulated as medicines.
We also regulate a fourth very limited type of goods known as other therapeutic goods (OTGs). These include sterilants, disinfectants, and tampons. Refer to Understanding regulation of disinfectants, sterilants and sanitary products and Tampons and menstrual cups.
Use of the term ‘investigational’ in this handbook is explained below:
- investigational product - any therapeutic good (including placebos) being tested or used as reference in a clinical trial
- investigational medicinal product - an investigational product that is a medicine
- investigational biological - an investigational product that is a biological
- investigational medical device - a medical device being assessed for safety or performance in a clinical investigation.
Types of therapeutic goods
There are four types of therapeutic goods.
Medicine: products that act by pharmacological, chemical, immunological, or metabolic means in or on the body of a human (includes biological medicines e.g. monoclonal antibodies, vaccines that do not contain human cells, plasma derivatives, recombinant products.
Medical Device: any instrument, apparatus, appliance, material or other article intended to be used for human beings for one of the following purposes:
- diagnosis, prevention, monitoring, treatment, or alleviation of disease
- diagnosis, monitoring, treatment, alleviation of, or compensation for an injury or handicap
- investigation, replacement or modification the anatomy or of a physiological process
- control of conception
and that does not achieve its principal intended action and then it's in or on the body by pharmacological, immunological, or metabolic means, but that may be assisted in its function by such means.
Biological: product made from or that contains human cells, human tissues, or live animal cells, tissues, or organs, that is used to:
- treat or prevent disease, ailment, defect or injury
- diagnose a condition of a person
- alter the physiological processes of a person
- test the susceptibility of a person to disease
- replace or modify a person's body parts.
(unless excluded or regulated as therapeutic goods, but not as biologicals)
Other therapeutic good: product that is not regulated specifically as a medicine, biological or medical device (includes sterilants, disinfectants, and tampons)
Refer to the Therapeutic Goods Act 1989 for the complete definitions.
Determine if the product is ‘unapproved’
Therapeutic goods must be included in the Australian Register of Therapeutic Goods (ARTG) before those goods can be lawfully imported into, exported from or supplied in Australia unless otherwise the subject of an exemption, approval or authority under the Therapeutic Goods Act 1989.
The therapeutic goods legislation provides a number of avenues to allow access to therapeutic goods that are not included in the ARTG. The CTN and CTA schemes provide for the lawful importation into and/or supply in Australia of ‘unapproved’ therapeutic goods for use solely for experimental purposes in humans.
For the purposes of this handbook, the reference to ‘unapproved’ therapeutic goods is an abbreviated expression1 which is intended to include:
- any medicine not included in the ARTG, such as any new formulation, strength or size, dosage form, name, indications, directions for use or type of container of a medicine already in the ARTG
- any medical device (including an in vitro diagnostic device (IVD)) not included in the ARTG, such as any new sponsor, manufacturer, device nomenclature system code, classification or unique product identifier (for certain classes of medical devices only) of a medical device already in the ARTG
- any in-house IVD medical device, used for the purpose of a clinical trial, where the laboratory providing the in-house IVD is unable to comply with the regulatory requirements for in-house IVDs (a laboratory developed test used for research purposes where results of such testing are not being used in patient diagnosis, treatment or management decisions would not be considered an in-house IVD)
- any biological not included in the ARTG such as:
- any new applicable standards, intended clinical use or principal manufacturer of a Class 1 or 2 biological already in the ARTG
- any new product name, dosage form, formulation or composition, therapeutic indication, type of container or principal manufacturer of a Class 3 or 4 biological already in the ARTG
- therapeutic goods already included in the ARTG to be used in a manner not covered by the existing entry in the ARTG.
The overall decision as to whether a CTN or CTA is required in relation to the use of the therapeutic goods is the responsibility of the trial sponsor. Consultation with the HREC that will approve the trial protocol may assist in the decision.
When a product is included in the ARTG, the entry applies to a particular sponsor (i.e. the individual or company intending to supply the goods). If a same or similar product is imported by another company or individual it is considered ‘unapproved’.
Trial sponsors who are considering supplying an ‘unapproved’ therapeutic good, need to recognise that it has not been evaluated by us for quality, safety, efficacy or performance.
Other pathways for accessing ‘unapproved’ therapeutic goods
The CTN and CTA schemes are not intended to be used as a means for obtaining access to ‘unapproved’ therapeutic goods for an individual patient. There are other mechanisms for lawfully supplying ‘unapproved’ therapeutic goods to individual patients outside clinical trials. Those mechanisms are:
- Special Access Scheme (SAS)
- Authorised Prescriber Scheme
- Personal Import Scheme.
Equally, the above pathways should not be used by health practitioners for the purposes of conducting a clinical trial. Health practitioners wanting to conduct a clinical trial (investigator-initiated trials) using an ‘unapproved’ therapeutic good, should do so using the CTN or CTA pathways, as appropriate.
The 'Authorised Prescriber' mechanism may be suitable to allow the lawful supply of the 'unapproved' therapeutic goods in a study that is not defined as a clinical trial. 'Clinical trials' are defined in our glossary. The TGA does not advise on study design or whether a study is defined as a 'clinical trial'. Sponsors should seek advice from the approving HREC.
In determining which pathway to use, it would also be advisable to consider the ethical acceptability of supplying an 'unapproved' therapeutic good outside the intent and purpose of each scheme.
The regulatory controls placed on clinical trials conducted through the CTN and CTA pathways provide sufficient assurance that high quality, credible data that contribute to the answering of specific scientific questions is collected, while also protecting the rights, safety and well-being of clinical trial participants.
Certain ‘unapproved’ therapeutic goods used in clinical trials
All ‘unapproved’ therapeutic goods, not just the investigational product(s), must be the subject of an exemption, approval or authority under the Therapeutic Goods Act 1989 to be lawfully supplied in a clinical trial.
Placebos
There is no entry for ‘placebo’ in the ARTG. Therefore, a placebo is considered to be an ‘unapproved’ therapeutic good and a CTN or CTA must be in place before a placebo can be supplied for use in a clinical trial. This is the case even if the investigational product(s) are already included in the ARTG.
Comparators
A comparator is an investigational or marketed product, or placebo, used as a reference in a clinical trial. If the trial sponsor determines a comparator to be an ‘unapproved’ therapeutic good, then a CTN or CTA must be in place before the product can be supplied for use in a clinical trial. This includes comparators that are included in the ARTG and altered for the purposes of a trial.
Medical software and mobile medical ‘apps’
Medical software and mobile medical ‘apps’ used in clinical trials may be regulated as medical devices and therefore may require exemption under the CTN scheme or approval under the CTA scheme. Further information can be found at Regulation of medical software and mobile medical ‘apps’.
Laboratory kits and procedure packs
If a laboratory kit or procedure pack (including any component or in its entirety) is determined by the trial sponsor to be an ‘unapproved’ medical device, then an exemption, approval or authority must be in place for the goods to be supplied (this includes the CTN and CTA schemes).
It is the trial sponsor’s decision based upon the advice from the approving HREC, on how to notify a procedure pack on the CTN form.
Non-investigational products used in clinical trials
Annex 13 of The Pharmaceutical Inspection Convention and Pharmaceutical Inspection Cooperation Scheme (jointly known as the PIC/S Guide to Good Manufacturing Practice for medicinal products) describes products other than the test product, placebo or comparator that may be supplied to participants in a trial.
Such products may be used as support medication for preventative, diagnostic or therapeutic reasons or needed to ensure that adequate medical care is provided for the participant (for example, an antiemetic supplied in a chemotherapy trial). If the trial sponsor determines such a product to be an ‘unapproved’ therapeutic good, then an exemption, approval or authority must be in place for the goods to be supplied (this includes the CTN and CTA schemes).
Genetically Modified Organisms (GMO)
Some therapeutic products may involve the use of a Genetically Modified Organism (GMO). Trials involving GMOs are subject to the Gene Technology Act 2000 and Gene Technology Regulations 2001, and corresponding state and territory legislation.
A trial sponsor proposing to use a GMO must apply to the Office of the Gene Technology Regulator (OGTR) for a licence or exemption documentation and this must be provided to the local Institutional Biosafety Committee at the clinical trial site.
Information on application procedures and approval is available from OGTR.
Also of relevance is our guidance Medicines produced by genetic manipulation.
The Australian regulatory environment
Clinical trials are regulated at a number of levels under Commonwealth and state and territory legislation in Australia.
The clinical trial environment in Australia is broad and there are various responsibilities resting with trial sponsors, HRECs, the approving authority (institution), investigators and Commonwealth and state and territory governments.
Australian regulatory requirements are specific to Australia. Trial sponsors who wish to conduct international multicentre trials are responsible for checking the legislative requirements in each country the trial is being conducted in (for example, trial sponsors should contact the U.S. Food and Drug Administration (FDA) for clinical trials conducted in the USA).
Therapeutic goods legislation
Therapeutic goods are regulated in Australia under the Therapeutic Goods Act 1989, the Therapeutic Goods Regulations 1990 and the Therapeutic Goods (Medical Devices) Regulations 2002. TGA is responsible for administering the Australian therapeutic goods legislation.
As regulatory requirements are amended from time to time, it is important to access the current official versions of the legislation. See About us for further information about our role in the regulation of therapeutic goods.
The therapeutic goods legislation relevant to the CTN and CTA schemes are outlined in the table below.
Legislative and regulatory provisions for the CTN and CTA schemes
| Therapeutic good | Legislation | CTN supply provisions | CTA supply provisions |
|---|---|---|---|
| Medicines and other therapeutic goods | Therapeutic Goods Act 1989 |
|
|
| Therapeutic Goods Regulations 1990 |
|
| |
| Fee provisions in Therapeutic Goods Regulations 1990 |
|
| |
| Information request provisions in Therapeutic Goods Act 1989 |
|
| |
| Medical devices | Therapeutic Goods Act 1989 |
|
|
| Therapeutic Goods (Medical Devices) Regulations 2002 |
|
| |
| Fee provisions in Therapeutic Goods (Medical Devices) Regulations 2002 |
|
| |
| Information request provisions in Therapeutic Goods Act 1989 |
|
| |
| Biologicals | Therapeutic Goods Act 1989 |
|
|
| Therapeutic Goods Regulations 1990 |
|
| |
| Fee provisions in Therapeutic Goods Regulations 1990 |
|
| |
| Information request provisions in Therapeutic Goods Act 1989 |
|
| |
| This table provides a guide to the Commonwealth therapeutic goods legislation related to the CTN and CTA schemes. Further information on state and territory legislation relevant to other aspects of clinical trial conduct can be found on the Australian Clinical Trials website. | |||
Declaration of Helsinki
The Declaration of Helsinki was used as the basis for developing guidance for good clinical practice in clinical trials. The principles of the Declaration of Helsinki also inform the ethical principles of the National Statement. However, the National Statement is the primary statement on ethical principles related to human research in Australia.
The National Statement on Ethical Conduct in Human Research
Regulation 12AD of the Therapeutic Goods Regulations 1990 and regulation 7.5 of the Therapeutic Goods (Medical Devices) Regulations 2002 provide that the use of therapeutic goods in a clinical trial must be in accordance with the National Statement.
The National Statement is developed jointly by the National Health and Medical Research Council (NHMRC) together with the Australian Research Council (ARC) and Universities Australia. It consists of a series of guidelines made in accordance with the National Health and Medical Research Council Act 1992 that are applicable to research with human participants. The National Statement is the Australian ethical standard against which all research involving humans, including clinical trials, are reviewed. It provides guidance to ensure any research involving humans is ethically sound.
In addition, the NHMRC, together with the ARC and Universities Australia, has also issued the Australian Code for the Responsible Conduct of Research. The purpose of this code is to guide institutions and researchers in responsible research practices. This is to ensure that their work has integrity and aligns with community expectations.
The National Statement on Ethical Conduct in Human Research
Regulation 12AD of the Therapeutic Goods Regulations 1990 and regulation 7.5 of the Therapeutic Goods (Medical Devices) Regulations 2002 provide that the use of therapeutic goods in a clinical trial must be in accordance with the National Statement.
The National Statement is developed jointly by the National Health and Medical Research Council (NHMRC) together with the Australian Research Council (ARC) and Universities Australia. It consists of a series of guidelines made in accordance with the National Health and Medical Research Council Act 1992 that are applicable to research with human participants.
The National Statement is the Australian ethical standard against which all research involving humans, including clinical trials, are reviewed. It provides guidance to ensure any research involving humans is ethically sound.
In addition, the NHMRC, together with the ARC and Universities Australia, has also issued the Australian Code for the Responsible Conduct of Research. The purpose of this code is to guide institutions and researchers in responsible research practices. This is to ensure that their work has integrity and aligns with community expectations.
Good Clinical Practice (GCP)
GCP is an international ethical and scientific quality standard for designing, conducting, recording and reporting clinical trials. Complying with GCP helps to ensure that the rights, safety and well-being of clinical trial participants are protected and that the trial data generated are credible. We recognise two internationally accepted GCP guidelines:
- for investigational medicinal products and investigational biologicals: International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) Guideline for Good Clinical Practice with TGA annotations
- for investigational medical devices: (ISO 14155).
For the purposes of this handbook the term 'Guideline for Good Clinical Practice' is used to reference the current version of the ICH document adopted by us with annotations.
Guideline for Good Clinical Practice
Regulation 12AD of the Therapeutic Goods Regulations 1990 specifies that the use of medicines and biologicals in a clinical trial must be in accordance with the Guideline for Good Clinical Practice.
The Guideline for Good Clinical Practice was developed by the ICH to provide a unified standard in the United States, the European Union and Japan to assist the mutual acceptance of clinical data by the regulatory authorities in these jurisdictions.
In order for data generated outside these regions to be accepted for regulatory submission, countries contributing data must ensure their clinical trials are conducted to principles equivalent to this guideline.
To ensure that Australia meets the high standards set out in this guideline, we have not produced a separate guideline for GCP. Instead, we have annotated the Guideline for Good Clinical Practice with TGA comments to ensure it reflects Australia’s specific regulatory and governance requirements.
Section 3 of the Guideline for Good Clinical Practice is not applicable to clinical trials in Australia. Instead, the responsibilities, composition, function, operations, procedural and record keeping requirements for HRECs in Australia are set out in the National Statement. If requirements specified in the National Statement appear to differ from those specified in the Guideline for Good Clinical Practice, the TGA recommends compliance with the National Statement.
ISO 14155
ISO 14155 has been developed by the International Organization for Standardization. It addresses GCP for the design, conduct, recording and reporting of clinical investigations carried out in human participants to assess the safety or performance of medical devices for regulatory purposes.
We recognise that ISO 14155 has substantially harmonised with the Guideline for Good Clinical Practice and therefore provides an equivalent standard. It also addresses the specific requirements of investigational medical device trials.
State and territory requirements
Each of the Australian states and territories has separate legislation relevant to the conduct of clinical trials. Investigators, HRECs, approving authorities and trial sponsors should ensure that they comply with the specific requirements for each jurisdiction.
National Mutual Acceptance (NMA) is a national system for mutual acceptance of scientific and ethical review of multi-centre clinical trials undertaken in publicly funded health services. It aims to enable the acceptance of single ethical and scientific review of human research projects in participating jurisdictions. Refer to the relevant state or territory health department for further information.
Research governance is the framework, systems and processes associated with the authorisation, commencement, conduct and monitoring of a clinical trial at a research site. Site authorisation is one part of research governance.
Individual jurisdictions have specific requirements as a part of their site-specific assessment and authorisation processes and these include additional compliance with data protection and radiation safety. Further information regarding research governance is available on the Australian Clinical Trials website.
Compliance with the relevant state and territory legislation for the manufacture, possession, supply or use of Scheduled medicines and poisons is also necessary. Scheduling is a national classification system that controls how medicines and poisons are made available to the public.
The Schedules are published in the Standard for the Uniform Scheduling of Medicines and Poisons (SUSMP) and are given legal effect through state and territory legislation.
The SUSMP is legally referred to as the Poisons Standard.
See Scheduling basics of medicines and chemicals in Australia for more information on the Poisons Standard.
The CTN and CTA schemes
The CTN and CTA schemes aim to provide considerable benefits by providing the momentum to research, developing new therapeutic goods locally and facilitating early patient access to new therapeutic developments.
An application or notification to conduct a clinical trial involving an ‘unapproved’ therapeutic good is independent of an application for product registration. We will accept a notification or application to conduct a clinical trial while an application for registration of the same product is under review. Similarly, we will accept an application for product registration while a clinical trial for the same product is under review or under way in Australia.
For the purposes of the CTN and CTA schemes, because of the potential for conflict of interest, the same person cannot be listed as the HREC contact and as the principal investigator, nor can the same person be listed as the clinical trial sponsor and the HREC contact.
We acknowledge that from time to time members of a HREC may be involved with a trial under review of that HREC. In these cases it is expected that the HREC will have procedures for managing conflicts of interest in accordance with the National Statement.
Choosing between the CTN and CTA schemes
The main difference between the CTN and CTA schemes is our level of involvement in reviewing data about the therapeutic goods before the clinical trial commences.
The choice of which scheme to use (CTN or CTA) lies firstly with the trial sponsor and then with the HREC that approves the protocol (except for certain Class 4 biologicals, which must be approved under the CTA scheme).
One of the determining factors for a HREC is whether the committee has access to appropriate scientific and technical expertise in order to assess the safety of the product. The approving authority takes the ultimate responsibility for determining whether the trial is allowed to proceed at the site.
If a HREC feels that it requires additional expertise to review a CTN, it may seek advice from external authorities or it may seek to collaborate with another HREC that has the required expertise.
A HREC may determine that it does not have access to the appropriate scientific and technical expertise to review the proposed trial under the CTN scheme and recommend review under the CTA scheme.
Medicines and biologicals
The CTN scheme may be used for earlier phase studies if there is adequate preclinical information available, especially regarding safety.
The CTA route is generally for high risk or novel treatments, such as gene therapy, where there is no or limited knowledge of safety. Under the biologicals framework, the CTA scheme is mandatory for a clinical trial of any Class 4 biological unless:
- evidence from previous clinical use supports the use of the biological. For example, the safety of the product has been evaluated in an earlier phase clinical trial. The effect on safety of changes in the manufacture of the product, or of use of the product for a new clinical indication, must be carefully considered; or
- a national regulatory body with comparable regulatory requirements has approved a clinical trial for an equivalent indication. Seek advice from us if you intend to use this provision. You will need to provide evidence that the safety review by the overseas regulator is equivalent to that which would be performed by us. In addition, the product used in the trial approved by the overseas regulator must be the same as that in the proposed trial. The effect on safety of changes in the manufacture of the product, or of use of the product for a new clinical indication, must be carefully considered.
- See the Australian regulatory guidelines for biologicals (ARGB) for further information and examples of Class 4 biologicals.
Medical devices
For medical device trials, the CTA scheme may be more appropriate where the experimental device introduces new technology, new material or a new treatment concept which has not been evaluated previously in clinical trials in any country.
The CTA scheme should also be considered for medical devices that pose a risk of serious patient harm when the HREC does not have, or have access to, adequate expertise in the preclinical technical areas.
This is especially relevant for implantable devices where areas of expertise include biological safety evaluation of medical devices, materials quality evaluation, engineering analysis of strength and fatigue of materials, finite element analysis for stress and fatigue prediction, and other computational simulations for device performance.
The CTN scheme
The CTN scheme is a notification scheme and as such we do not review or evaluate any data relating to clinical trials at the time of submission. All material relating to the proposed trial, including the trial protocol is submitted directly to the HREC.
The HREC is responsible for assessing the scientific validity of the trial design, the balance of risk versus harm of the therapeutic good(s) and the overall ethical acceptability of the trial.
We may request further information from the trial sponsor regarding a clinical trial submitted to us under the CTN scheme (see Requests for information).
We may ask for information such as the investigator’s brochure and protocol for first-in-human trials or because we have a degree of concern. The specific reasons will be different for each trial. The request will be made under the appropriate section of the Therapeutic Goods Act 1989.
Once we have reviewed this information, we may ask you specific questions to address any deficiencies or concerns that we identify.
A clinical trial must be notified to us using the online CTN form before the trial sponsor can supply the ‘unapproved’ therapeutic goods in the trial.
A clinical trial is deemed to be notified as soon as the CTN form has been submitted and the relevant fee has been paid (as set out in item 3 of Schedule 5A to the Therapeutic Goods Regulations 1990 or item 2.3 of Schedule 4 to the Therapeutic Goods (Medical Devices) Regulations 2002).
Once this occurs the exemption comes into effect and the trial sponsor can supply the goods subject to the necessary HREC approval and institutional authorisations.
The CTN exemption allows for the lawful supply of ‘unapproved’ therapeutic goods in a clinical trial. However, it is the responsibility of the trial sponsor to ensure that all relevant approvals (for example, ethics approval and site authorisation) are in place before commencement of the trial and supplying the ‘unapproved’ therapeutic goods.
If a therapeutic good is included in the ARTG during the course of the clinical trial then the trial sponsor must consult the HREC to determine the ethical acceptability of continuing to supply an ‘unapproved’ version.
Guidance on the online process for CTNs is available on our website.
Variations to a CTN
If there are any changes to the trial details notified to us (such as a change in the details of the principal investigator, change in address of the site or a change in the therapeutic good) it is necessary for the trial sponsor to update the relevant fields on the online CTN form. Certain changes to a previously notified CTN will incur a fee (see our Clinical trials FAQ for further information).
Notification of completion of a clinical trial under a CTN
The purpose of the CTN scheme is to provide an exemption for the supply of ‘unapproved’ therapeutic good in a clinical trial. Therefore, the completion advice should be submitted to us once the clinical trial related activity afforded by this exemption is complete and the exemption would no longer be required.
Trial sponsors should only notify us of the completion of a clinical trial after the trial has been completed at all sites. It is not necessary to notify completion dates for individual trial sites. This would usually correspond with the last patient last visit; however, it the responsibility of the trial sponsor to determine when the exemption is no longer required. Trial sponsors should notify us when a trial is completed using the online CTN form.
To ensure we maintain an accurate record for each clinical trial, we must be notified of:
- the date the clinical trial was completed. This is the last date of completion for all sites. It is not necessary to notify completion dates for individual sites
- the reason the clinical trial ceased, for example, concluded normally, insufficient recruits, etc.
We do not require trial sponsors to submit end of trial study reports. However, this information should be made available if we request it.
The CTA scheme
The CTA scheme is an evaluation process and involves the review by us of relevant, but limited, scientific data (which may be preclinical and early clinical data) prior to the start of a trial. This can be particularly useful for therapeutic goods that are in the early stages of development.
Our primary responsibility is to review the safety of the product and the HREC is responsible for considering the scientific and ethical issues of the proposed trial protocol. Conduct of a clinical trial under the CTA scheme must also be approved by the responsible HREC(s).
The trial sponsor's interaction with us for trials conducted under a CTA involves the submission of a formal CTA application.
If a trial sponsor is considering submitting a CTA application, we strongly encourage them to contact us for pre-submission advice regarding the application process.
CTA forms
CTA applications are submitted using paper-based forms. There are two forms (part 1 and part 2) that must be completed by the sponsor and submitted to us via clinical.trials@health.gov.au.
Part 1 is the formal CTA application. The Part 1 form must be completed by the sponsor of the trial and submitted to us together with the data for evaluation. The CTA application form should be emailed to us a clinical.trials@health.gov.au. Supporting data for the CTA application should be provided in electronic format. It is preferable for data to be sent on USB or CD-ROM via post. The appropriate fee must be paid.
The Part 2 form is used to notify us of the commencement of each new trial conducted as per the usage guidelines approved in the CTA application. There is no fee for notification of commencement of the trial.
Variation to a CTA
The evaluation of a CTA application includes consideration of the manufacturing and quality and safety data in conjunction with the trial’s usage guidelines, to inform a risk-benefit decision by TGA on whether or not to approve the clinical trial.
Any significant changes to the information provided in support of the trial are considered a ‘variation’ and need to be approved as they have the potential to affect the initial decision to approve a trial.
We advise clinical trial sponsors to contact us via clinical.trials@health.gov.au if they intend to vary a previously approved CTA application.
Notification of a trial completion under a CTA
Trial sponsors should notify us of trial completion using the CTA clinical trial completion advice form. Trial sponsors should only notify us of the completion of a clinical trial after the trial has been completed at all sites. It is not necessary to notify completion dates for individual trial sites. This would usually correspond with the last patient last visit.
Extensions of clinical trials
The trial sponsor may seek approval from a HREC to conduct further trials with the same product. In each case, the trial sponsor is required to submit a CTN or CTA to us for trials involving the use of ‘unapproved’ therapeutic goods.
Building in a trial extension provision into the original protocol design not only fulfils GCP requirements but also can allow treatment to continue without having to prepare another trial proposal after the initial trial ceases. Of course, this is not the only way to provide ongoing treatment post-trial.
Post-trial provisions
The National Statement requires a HREC to be satisfied that it is clearly explained to participants whether they will have continued access to treatments received during the trial when the trial is completed.
All parties involved in the planning, approval and conduct of clinical trials that involve the use of ‘unapproved’ therapeutic goods should consider mechanisms for allowing participants continued access to those goods following completion of the trial.
This is especially important for participants where treatment has been found to be effective and where long term therapy would be appropriate. Consideration should be given to including a post study supply component in the trial protocol.
If supply of clinical trials goods is to continue after completion of the clinical trial, then it must be supplied within the provisions of the Therapeutic Goods Act 1989 for access to ‘unapproved’ therapeutic goods. If additional safety data is to be collected, the continuation of supply is usually undertaken using an open-label extension study. An open-label extension study is usually notified as a separate trial under the CTN and CTA schemes.
Alternatively, one of the other mechanisms for accessing ‘unapproved’ goods may be appropriate, such as the Special Access Scheme, Authorised Prescriber, or Personal Import Scheme.
Responsibilities under the CTN and CTA schemes
Clinical trials are conducted by researchers as part of a broad group of stakeholders with varying responsibilities under the therapeutic goods legislation. These include trial sponsors, HRECs, approving authorities (institution in which the trial is being conducted), investigators and TGA.
The key responsibilities for each of these stakeholder groups in the context of the CTN and CTA schemes are described below. There are other responsibilities for each stakeholder that are outside of TGA requirements.
The Australian Clinical Trials website provides further information on other requirements.
Role of trial sponsors
Sponsor definition
All clinical trials conducted in Australia must have a trial sponsor that is an Australian entity (an overseas company cannot be the sponsor of a trial in Australia). Sponsors of trials under the CTN or CTA schemes may include individuals, companies, institutions, or organisations. Within the context of this handbook, we recognise two distinct definitions for the term 'sponsor':
- sponsor in relation to therapeutic goods: as defined in Section 3 of the Therapeutic Goods Act 1989
- sponsor in relation to clinical trials: as defined in the Guideline for Good Clinical Practice for medicines or biologicals or in ISO 14155 for medical devices
The trial sponsor is responsible for the initiation, management and financing (or arranging the financing) of the trial and carries the medico-legal responsibility associated with its conduct. The Australian trial sponsor is also the entity that is responsible for submitting a CTN or CTA to us.
Trial sponsors and the therapeutic goods legislation
The therapeutic goods legislation, as referenced in Legislative and regulatory provisions for the CTN and CTA schemes, specifies responsibilities of the trial sponsor under the CTN and CTA schemes. The trial sponsor takes the overall responsibility for trials conducted under the CTN and CTA schemes.
Trial sponsors should be aware of their responsibilities under the therapeutic goods legislation. Under the CTN scheme, the approval of the goods for use in the trial must be given by the sponsor (if the sponsor is conducting the trial), or by the body or organisation conducting the trial for the sponsor, having regard to the advice of the ethics committee responsible for monitoring the conduct of the trial. The sponsor must not receive advice from the ethics committee that is inconsistent with the continuation of the trial.
Under the CTN and CTA schemes the use of therapeutic goods in the trial must be in accordance with the Guideline for Good Clinical Practice, the National Statement and the protocol approved by the HREC responsible for monitoring the conduct of the trial.
The trial sponsor must also comply with the requirements of any other Commonwealth and/or state and territory legislation in relation to clinical trials and the supply of therapeutic goods.
Investigator-initiated trials
The requirement to conduct a trial in accordance with GCP and all other regulatory requirements does not depend on whether the trial sponsor is a commercial company or a non-commercial institution or organisation (for example, a hospital or university).
Where an investigator initiates and organises a trial without the involvement of an institution, he or she will take on the role of the trial sponsor and will then be responsible for the extensive GCP and regulatory requirements associated with both the management and conduct of the trial.
Where another party, such as a pharmaceutical or biotechnology company, provides investigational product (or other support) for an investigator-led CTN or CTA trial, the provision of that support does not oblige the company to take the role of trial sponsor.
Trial sponsor oversight of delegated functions
The Guideline for Good Clinical Practice and ISO 14155 allows a trial sponsor to delegate any or all of the sponsor's trial-related duties and functions, including safety reporting, to a third party such as a contract research organisation.
However, the ultimate responsibility for the quality and integrity of the clinical trial data resides with the trial sponsor. The trial sponsor retains overall responsibility for all delegated functions in accordance with the Guideline for Good Clinical Practice and ISO 14155.
This also applies when a non-commercial trial sponsor delegates duties; for example, to the coordinating principal investigator or trial coordinating centre.
The contracts and agreements between trial sponsors and third parties must ensure all roles and responsibilities are clearly defined. The approving HREC retains discretion to review and approve proposed delegations of trial sponsor functions.
Where sponsor functions are delegated to third parties, the trial sponsor should maintain appropriate oversight of those functions.
Reporting a change or transfer of trial sponsor
The transfer or change of trial sponsorship requires the submission of a new CTN or CTA form by the new trial sponsor for each of the clinical trials involved. This will ensure that the exemption or approval to supply the ‘unapproved’ therapeutic goods remains valid.
We recommend that the clinical trial start date reflects the date the new trial sponsor assumes responsibility for the clinical trial(s). Generally, we have no objection to clinical trials continuing while all the necessary endorsements are obtained provided the original sponsor is able to fulfil their obligations until the new sponsor assumes responsibility.
The sponsor relinquishing sponsorship may notify the TGA that the exemption is no longer required by submitting a completion advice to us.
Trial sponsor responsibility of trial management and monitoring
The responsibilities of the trial sponsor are extensive; however, this handbook does not describe all sponsor responsibilities as they are clearly described in Section 5 of the Guideline for Good Clinical Practice and throughout ISO 14155.
Instead, this section provides detail on the overarching responsibility of the trial sponsor for monitoring and managing their trials; particularly the responsibilities for safety reporting to us. Trial sponsors should ensure that a trial is appropriately monitored for compliance with GCP.
The quality of information generated when clinical trials are conducted impacts on the future care of the Australian population. Before initiating a trial, the trial sponsor should ensure that quality management systems are in place and that these systems are robust enough to fulfil all GCP and regulatory requirements, including relevant state and territory legislation.
For example, when designing a trial, the Guideline for Good Clinical Practice requires trial sponsors to use a multi-disciplinary team of qualified individuals (for example, biostatisticians, clinical pharmacologists, and physicians) as appropriate, throughout all stages of the trial process, from designing the protocol and case report forms to analysing and preparing interim and final clinical trial reports.
When planning a clinical trial, trial sponsors should have processes in place to ensure the risks associated with its conduct are identified and assessed so that adequate trial monitoring and management plans can be developed to mitigate risk that may adversely impact on trial quality or participant safety.
We recognise that clinical trials under the CTN or CTA schemes vary greatly in terms of the risks posed to participants when compared to normal clinical care. The NHMRC guidance on Risk-based Management and Monitoring of Clinical Trials Involving Therapeutic Goods provides further information on the implementation of risk based management and monitoring plans.
Conducting ongoing safety evaluation and safety reporting
The Guideline for Good Clinical Practice and ISO 14155 places the responsibility for the ongoing safety evaluation of the investigational product used in a clinical trial, with the trial sponsor.
To ensure there is appropriate safety oversight, trial sponsors should generally use an independent committee or independent individuals. A Data Safety Monitoring Board (DSMB) may be convened based on the potential risks and benefits to participants associated with the trial and the trial design. The NHMRC guidance on Data Safety Monitoring Boards (DSMBs) provides advice on the use of DSMBs and also describes other safety monitoring structures that may be utilised when a DSMB is not warranted.
See Safety reporting to TGA for CTN and CTA trials for information on how trial sponsors should notify us of safety reports for CTN and CTA trials.
Record keeping obligations
The trial sponsor’s record keeping obligations are outlined in section 5.5 of the Guideline for Good Clinical Practice (with our annotations for local context) and section 7.4 of ISO 14155.
There may be longer (or indefinite) retention periods for certain trials (for example, trials involving children and trials involving gene therapy). State and territory and institutional requirements may also specify longer retention periods.
Registration of trial
The National Statement obligates researchers to ensure that their trials are registered in a publicly accessible database before recruitment of the first participant. The International Committee of Medical Journal Editors (ICMJE) has a policy requiring trial registration prior to publication.
The Australian Clinical Trials website describes some of the reasons why trial registration is important and provides a link to appropriate registries.
The Australian Code for the Responsible Conduct of Research states, ‘researchers have a responsibility to their colleagues and the wider community to disseminate a full account of their research as broadly as possible’. The Code also describes responsibilities in relation to the dissemination of research findings.
Notifying trial sites
While a number of sites may be involved in trial related activities, the need to notify each site to us on the CTN and CTA forms is a separate matter. The trial sponsor should consult the approving HREC to determine which sites to notify to us.
However, the essential documents relating to a trial are expected to describe in detail where each trial related activity will be taking place. These documents as well as any relevant legal arrangements between a trial sponsor and service provider should be made available to us if required.
Role of Human Research Ethics Committees (HRECs)
We have adopted a risk-based approach to the governance of clinical trials in Australia, a concept that is supported by the Organisation for Economic Cooperation and Development (OECD) in their Recommendation on the Governance of Clinical Trials.
We recognise that Australian HRECs have the mission and competence to oversee clinical trials under such a framework. As a result, HRECs play a central role in reviewing clinical trials that are subject to the therapeutic goods legislation.
The National Statement requires that all clinical trials must be reviewed by a HREC. The responsibilities, composition, function, operations, procedural and record keeping requirements for HRECs in Australia are set out in the National Statement. Therefore, Section 3 of the Guideline for Good Clinical Practice is not applicable to clinical trials in Australia.
If requirements specified in the National Statement appear to differ from those specified in the Guideline for Good Clinical Practice, the TGA recommends compliance with the National Statement.
HRECs also need to be aware of relevant state and territory laws pertaining to clinical trials and the supply of therapeutic goods. HRECs have a high level of independence and are responsible for establishing their own processes for receiving and reviewing research proposals.
Recent reforms under national and state and territory programs to harmonise HREC processes and to minimise duplication of review of multicentre research projects has resulted in increased standardisation of process.
The NHMRC’s National Approach to Single Ethical Review of Multi-Centre Research (National Approach) aims to enable the recognition of a single ethics and scientific review of multi-centre human research within or across Australian jurisdictions. A key component of the National Approach is the National Certification Scheme of Institutional Processes related to the Ethical Review of Multi-centre research (National Certification Scheme).
Under the National Certification Scheme, some institutions have had their ethical review processes assessed by the NHMRC against an agreed set of national criteria. This has allowed HRECs to demonstrate, through assessment, that they are following the relevant guidelines and legislation when reviewing trial protocols and have processes consistent with the expectations of the National Statement.
A list of institutions with certified review processes is available on the NHMRC website.
NHMRC has also developed the Human Research Ethics Application (HREA) as a national ethics form. Trial sponsors and researchers should use the HREA unless advised otherwise.
HRECs and the therapeutic goods legislation
The therapeutic goods legislation sets out a number of requirements for HRECs under the CTN and CTA schemes (See Legislative and regulatory provisions for the CTN and CTA schemes for specific references to the legislation). Under the Act, an ethics committee means a committee:
- constituted and operating as an ethics committee in accordance with guidelines issued by the CEO of the National Health and Medical Research Council as in force from time to time; and
- which has notified its existence to the Australian Health Ethics Committee (AHEC) established under the National Health and Medical Research Council Act 1992.
A list of HRECs registered with NHMRC can be found on the NHMRC website.
Under the CTN and CTA schemes, the use of the therapeutic goods in a clinical trial must be in accordance with the Guideline for Good Clinical Practice, the National Statement and the trial protocol approved by the HREC that has the function of monitoring the conduct of the trial (see The HRECs’ role in monitoring a clinical trial).
Therefore, HRECs are responsible for approval of the trial protocol under both the CTN and CTA schemes. The HREC and the institution are responsible for establishing what information should be provided in support of an application and how the application will be reviewed by the committee.
The HREC should request any additional information that it believes is necessary to undertake review of the proposed research.
The HRECs’ role in monitoring a clinical trial
In approving a clinical trial protocol under the CTN and CTA schemes, the HREC accepts responsibility for ‘monitoring’ the conduct of the trial.
The word ‘monitoring’ in this context is used to describe the continuing review and oversight of the trial by the HREC. For example, this may include receipt of progress reports, safety information, approval of protocol amendments, notifications of serious breaches of the protocol or good clinical practice and updated trial documentation.
As the trial progresses, the HREC is also responsible for monitoring the continued benefit-risk ratio of the trial; in particular, whether any safety reports they receive impact on the continued ethical acceptability of the trial.
For further information on the safety monitoring and reporting responsibilities of researchers, trial sponsors, HRECs and institutions see the NHMRC Guidance: Safety Monitoring and Reporting in Clinical Trials Involving Therapeutic Goods.
The trial sponsor’s monitoring responsibilities are outlined in both the Guideline for Good Clinical Practice and ISO 14155 and discussed further under Role of trial sponsors.
Withdrawal of approval by a HREC
It is a condition of both the CTN and CTA schemes that the use of the therapeutic goods in a clinical trial must cease if the HREC informs the principal investigator that the use is inconsistent with the protocol or a condition of approval by the HREC.
In addition, one of the conditions under which therapeutic goods are supplied in a CTN trial is that the trial sponsor must not receive advice from the HREC that is inconsistent with continuing the trial.
The HREC may review its ethics approval at any time in the light of safety reports, progress reports, issues raised by media reports or any other information (for example, a report of serious breach) that raises serious safety concerns or concerns about the conduct of the trial.
If the HREC becomes aware of information that may significantly impact on the rights, safety or wellbeing of participants or the credibility of trial data, it may advise the trial sponsor, approving authority and principal investigator that it intends to withdraw ethical approval at one or more sites for which the HREC is responsible.
If the trial sponsor is not able to allay these concerns, then the HREC may withdraw ethical approval. The HREC should advise the trial sponsor, principal investigator, and the relevant approving authorities of any decision to withdraw approval. We should also be notified of the decision to withdraw ethics approval for a trial conducted under the CTN or CTA schemes.
The National Statement outlines the circumstances when research should be discontinued. The NHMRC has also published guidance on the Reporting of Serious Breaches of Good Clinical Practice (GCP) or the Protocol for Trials Involving Therapeutic Goods which provides further information.
The continued supply of the ‘unapproved’ therapeutic goods would be unlawful if a HREC suspends or withdraws ethics approval for a trial conducted under the CTN or CTA scheme.
Circumstances that may lead to a HREC withdrawing its approval for a trial are most likely to be identified in the course of monitoring a trial. These may include:
- evidence of significant or repeated deviation from the trial protocol and that, as a result, the welfare and rights of participants are not or will not be protected
- evidence that allowing the trial to continue carries an unacceptable risk of death, serious illness or serious injury to trial participants
- evidence from progressive review of a comparative study shows that one treatment proves to be so much better or worse that to continue the trial would disadvantage one group of participants
- evidence that the conduct of the trial is in breach of Commonwealth, state or territory laws.
Role of approving authorities
Approving authorities are public or private legal entities (institutions or organisations) where trials are conducted (i.e. trial sites).
For investigator-led trials, the institution that is the approving authority may also be the trial sponsor. In such cases the institution must ensure that its overarching quality management systems delineate its responsibilities as a trial sponsor from its responsibilities as a trial site.
Private clinics may be used as trial sites for clinical trial purposes and therefore take on the responsibilities of the approving authority. These sites must comply with good clinical practice and all other applicable clinical trial legislation and requirements.
Approving authorities conducting trials under the CTA and CTN schemes have specific responsibilities as outlined in the National Statement, Guideline for Good Clinical Practice and ISO 14155 and the Therapeutic Goods Act 1989.
Approving authorities and the therapeutic goods legislation
Under the CTN and CTA schemes, the approving authority gives the final authorisation for the conduct of the trial at the site following approval by the reviewing HREC (see Legislative and regulatory provisions for the CTN and CTA schemes for specific references to the legislation).
Site authorisation
We do not have a role in the site assessment and authorisation processes, this is the responsibility of the approving authority.
The Australian Code for the Responsible Conduct of Research states that bodies responsible for research should:
- ‘provide an appropriate research governance framework through which research is assessed for quality, safety, privacy, risk management, financial management and ethical acceptability’.
These operations are generally overseen by a research office within the institution.
The research office’s responsibilities are distinct from those of the HREC. The granting of ethical approval by a HREC does not oblige an approving authority to grant authorisation at their site as the site may not have the capacity or capability to complete the trial based on protocol requirements.
As part of the process to confirm whether authorisation should be granted, the research office liaises with the site principal investigator and the trial sponsor to establish requirements to grant site authorisation.
Monitoring by the approving authority
The National Statement sets out obligations relevant to monitoring for institutions. Monitoring under the National Statement refers to the process of verifying that the conduct of research conforms to the approved proposal. Responsibility for ensuring that research is reliably monitored lies with the institution under which the research is conducted.
Approving authorities must be made aware of any issues that may impact on trial authorisation or institutional risk (for example, significant safety issues identified by the trial sponsor). The approving authority may withdraw authorisation if necessary. Such a withdrawal should be communicated to the HREC and to us.
Where a HREC has withdrawn ethical approval, the approving authority is obliged to ensure that the principal investigator promptly suspends the research. Arrangements should also be made to meet the needs of the participants in the clinical trial.
Role of principal investigators
The principal investigator is referenced in the therapeutic goods legislation relevant to the CTN and CTA schemes (see Legislative and regulatory provisions for the CTN and CTA schemes for specific references to the legislation).
Investigators are responsible for protecting the rights and safety of trial participants under their care. A full list of investigator responsibilities can be found in the Guideline for Good Clinical Practice or ISO 14155. The principal investigator is the person responsible, individually or as a leader of the research team at a site, for the conduct of a clinical trial at that site. As such, the principal investigator is responsible for adequately supervising his or her research team.
The principal investigator must conduct the clinical trial in accordance with the clinical trial protocol. Any significant departures that impact on the safety of participants or the reliability of trial data that occur at their site should be reported to the trial sponsor, HREC and approving authority, in line with NHMRC guidance on the Reporting of Serious Breaches of Good Clinical Practice (GCP) or the Protocol for Trials Involving Therapeutic Goods.
The principal investigator must ensure adequate medical cover is provided for the trial and ensure that all adverse events are reported in accordance with the trial protocol. The NHMRC Guidance: Safety monitoring and reporting in clinical trials involving therapeutic goods provides more information on the safety reporting responsibilities of investigators.
Where an investigator initiates and organises a trial, he or she will take on the role of the trial sponsor and will then be responsible for the GCP and regulatory requirements associated with both the management and conduct of the trial.
Role of TGA
Requests for information
Under the Therapeutic Goods Act 1989, we can request certain information or documents, in writing, about therapeutic goods exempt under the CTN scheme or approved under the CTA scheme relating to:
- the supply of the goods
- the handling of the goods
- the monitoring of the supply of the goods
- the results of the supply of the goods.
This can include documentation such as the investigator’s brochure and protocol, further information about safety reports, clarification about the safety profile of a specific therapeutic good, or details of problems or complaints.
We can require the trial sponsor of the goods exempt under the CTN scheme or the person who is granted approval under the CTA scheme to provide this information or documents.
It is a criminal offence to fail to comply with a request for information or documents that is made under the legislation, or to knowingly provide false or misleading information or omit any matter without which the information is misleading.
Civil penalties also apply for giving false or misleading information or documents. See Legislative and regulatory provisions for the CTN and CTA schemes for the relevant sections of the Therapeutic Goods Act 1989 relating to requests for information.
With respect to the CTA scheme, we can inspect clinical trial sites as described in regulation 12AC of the Therapeutic Goods Regulations 1990 and regulation 7.4 of the Therapeutic Goods (Medical Devices) Regulations 2002. We can also request the principal investigator to answer questions and produce any records or documents we request.
We can release information obtained in a response to a request for information to an authority of the Commonwealth, a state or a territory that has functions relating to therapeutic goods as well as medical boards.
When further action is required
We may ask further questions or raise concerns that may need to be addressed from the review of requested information or documents. Further action may be required if any issues of concern are identified. This may include liaison with trial sponsors, principal investigator(s), approving authorities or the approving HREC.
Therapeutic goods supplied under the CTN scheme are exempt from the requirement to be included in the ARTG, subject to conditions. If these conditions are not complied with then the goods are no longer exempt from inclusion in the ARTG and cannot be lawfully supplied. Offence provisions may also apply where a person does an act, or omits to do an act, that results in a breach of condition of a CTN exemption.
Under the CTN scheme, if we direct the trial not be conducted or become aware that to conduct or continue the trial would be contrary to the public interest then the goods used in the trial would no longer be exempt from inclusion in the ARTG. For example, this may be where it comes to our notice that allowing the trial to proceed or continue carries an unacceptable risk of death, serious illness or serious injury.
We can also revoke an approval of a clinical trial under the CTA scheme where the conditions of approval are not met. In addition to revoking approval, offence provisions may also apply where goods which are the subject of a CTA approval are not used in accordance with that approval or a statutory condition made under subsection 19(4A) of the Therapeutic Goods Act 1989.
Procedures following the revocation of approval under the CTA scheme or a breach of the conditions of the CTN scheme would be determined on a case-by-case basis based on the impact on participants and their ongoing safety. It is the trial sponsor’s responsibility to notify the HREC of a change in CTN or CTA status; however, we may liaise with the HREC in certain circumstances in a confidential manner.
For multicentre studies the trial sponsor will have the additional responsibility of notifying all sites. A lead HREC in a multicentre study will need to liaise with the sites and trial sponsor when determining which, if any, are affected and the actions they need to apply.
Criminal and civil penalties may apply where a therapeutic good is used in a clinical trial in circumstances where a CTN exemption or a CTA approval no longer applies to the supply of the therapeutic good.
A CTN exemption will automatically cease to apply to the supply of a therapeutic good in a clinical trial if a condition of the exemption is breached.
A CTA approval will cease to apply to the supply of a therapeutic good in a clinical trial if we revoke the approval. We may revoke approval if a trial sponsor breaches a condition of approval.
Offence provisions may apply where there is a breach of a condition of a CTN exemption or a CTA approval.
Release of information
Information provided to us concerning the use of ‘unapproved’ therapeutic goods in relation to clinical trials will be treated as confidential. For information about the release of this information, see TGA approach to disclosure of commercially confidential information.
We have powers under the Therapeutic Goods Act 1989 to release certain information about therapeutic goods in specified circumstances. One such circumstance is where release is necessary to ensure the safe use of particular therapeutic goods. Depending on the circumstances, there may be a need to release information to a HREC where release is necessary to ensure the safe use of therapeutic goods in a clinical trial.
Concerns or complaints about a clinical trial
A person or organisation may report a perceived breach or questionable practice involving the use of an 'unapproved' therapeutic good in a clinical trial anonymously through our website or contact us for assistance.
Safety reporting to TGA for CTN and CTA trials
Trial sponsors should refer to the NHMRC Guidance: Safety Monitoring and Reporting in Clinical Trials Involving Therapeutic Goods (NHMRC Guidance) for safety reporting requirements. The NHMRC guidance addresses the monitoring, collection and reporting of adverse events that occur in clinical trials involving therapeutic goods conducted under the CTN or CTA schemes. The NHMRC Guidance has aligned with the European Union’s Clinical Trial Regulations: Regulation EU No 536/2014.
The NHMRC Guidance outlines the responsibilities of trial sponsors, investigators, HRECs and institutions (referred to as the approving authority in this handbook); however, it is the trial sponsor that is responsible for reporting to us.
We have adopted the CPMP/ICH/377/95 Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting in principle with annotations for local regulatory requirements. For clinical trials, we now reference the European Commission terms for categorising adverse events as they are more widely used in Australia.
For example, the term 'Suspected Unexpected Serious Adverse Reaction' (SUSAR) has replaced the term 'Serious and Unexpected Adverse Drug Reaction'.
The term ‘investigational medicinal product’ in the NHMRC Guidance applies to both medicines and biologicals.
In this handbook, medicines and biologicals are referenced separately so that the differing regulatory requirements can be highlighted.
We have aligned the definitions for all safety reporting with those provided in the NHMRC Guidance.
The NHMRC Guidance outlines the trial sponsor’s safety reporting responsibilities for CTN and CTA trials. We have provided further information on how the trial sponsor should notify us of all relevant safety reports for CTN and CTA trials in the following tables.
Safety reporting timeframes for CTN and CTA trials
Single case events from Australian sites: SUSARS and USADEs
| Type of event | Type of good | Report format | Timeframe |
|---|---|---|---|
| Suspected unexpected serious adverse reactions (SUSARs) from Australian sites only | Medicines and biologicals | The new Electronic Data Interchange (EDI) functionality, allows sponsors to submit adverse event reports directly from their system to us. Review the Electronic submission of individual case safety reports link regarding this functionality. If you require assistance in connecting to this service, send email to adr.reports@health.gov.au OR
Visit Adverse Event Management System (AEMS) for more information about reporting to us. |
|
| Unanticipated serious adverse device effects (USADEs) from Australian sites only | Medical devices |
|
|
Significant safety issues* and overseas regulatory action
| Type of event | Type of good | Report format | Timeframe |
|---|---|---|---|
| Significant safety issues (SSIs) requiring implementation of urgent safety measures (USMs) | All therapeutic goods | In writing to the Pharmacovigilance Branch via email to clinical.trials@health.gov.au. | Within 24 hours (where possible) and in any case, no later than 72 hours of the measure being taken. |
| Action with respect to safety that has been taken by another country's regulatory agency (relevant to an ongoing clinical trial in Australia) | All therapeutic goods | In writing to the Pharmacovigilance Branch via email to clinical.trials@health.gov.au. | Without undue delay and no later than 72 hours of the trial sponsor becoming aware of the action. |
All other significant safety issues (SSIs): Notification of an amendment** Temporary halt of a trial for safety reasons Early termination of a trial for safety reasons | All therapeutic goods | In writing to the Pharmacovigilance Branch via email to clinical.trials@health.gov.au. | Without undue delay and no later than 15 calendar days of the trial sponsor becoming aware of the issue or temporary halt or early termination. |
*SSIs that arise from analysis of overseas reports (relating to a clinical trial in Australia) should be reported to us as per the timeframes above.
** We should receive notification that a SSI has occurred but the amendment revising trial documentation should be submitted to the HREC only.
Note: A SUSAR or USADE may also meet the definition of an SSI.
Other report types
| Type of event | Type of good | Report format | Timeframe |
|---|---|---|---|
| Other single case adverse events (AEs) | All therapeutic goods | Up to date tabulations or line listings | On our request |
| Annual safety reports | All therapeutic goods | Development Safety Update Reports (DSURs) or other annual safety reports | On our request |
Single case events from Australian sites: SUSARs and USADEs
Individual SUSARs and USADEs from Australian sites must be reported to us. Refer to the NHMRC Guidance for definitions of SUSARs and USADEs.
Even if initial information is limited (i.e. less than the minimum information required for expedited reporting as outlined in the CPMP/ICH/377/95 Guideline for Clinical Safety Data Management: Definitions and Standards for Expedited Reporting) these details should still be forwarded to us pending receipt and provision of further data.
Managing blinded trials
The blind should be broken with care when trial sponsors are reporting SUSARs and USADEs to us.
We recommend that the trial sponsor breaks trial code for a particular patient, even if the investigator has not unblinded the case. This may necessitate withdrawal of the subject from the trial.
It is essential that unblinded information is only accessible to sponsor staff who need to be involved in the reporting of these events to us. The blind should be maintained for persons responsible for the ongoing conduct of the trial, particularly those who are responsible for data analysis and interpretation of results.
Breaking a trial code may also be required in the interests of public safety. However, this is a serious undertaking that has the capacity to invalidate the entire clinical trial. Trial sponsors, HRECs and DSMBs are invited to consult with us if this situation arises.
We recognise that in some trials (for example, stroke trials) efficacy end-points such as mortality or serious morbidity could also be SUSARs or USADEs. As such, the trial’s integrity may be compromised if the blind is systematically broken. These outcomes would be considered to be 'disease-related' and exempted from expedited reporting. However, where it becomes apparent from the trial sponsor's own monitoring and analysis of data that the number of cases of a fatal or serious nature is in excess of that which could reasonably be expected, trial sponsors should reconsider the issue of blinding and report this information to us.
The Note for Guidance on Clinical Safety Data Management: Definitions and Standards for Expedited Reporting provides further information on managing blinded therapy cases.
For trials in which efficacy end-points could also be SUSARs or USADEs, the trial protocol should state explicitly when the trial will be unblinded because of safety concerns.
Reporting of overdoses and interactions between therapeutic goods
Trial sponsors should report SUSARs that occur as a result of cases of accidental or intentional overdose. This includes, for example, instances where the investigational medicinal product is suspected of causing suicidal ideation or medication errors leading to an accidental overdose. There is no requirement to report overdoses with no associated adverse reaction as SUSARs.
Any SUSAR or USADE that results from interactions between therapeutic goods should also be reported to us.
Refer to the NHMRC Guidance: Safety Monitoring and Reporting in Clinical Trials Involving Therapeutic Goods for safety reporting requirements for other stakeholders.
Sponsor reporting of SUSARs and USADEs to TGA (for trials conducted under the CTN or CTA schemes)
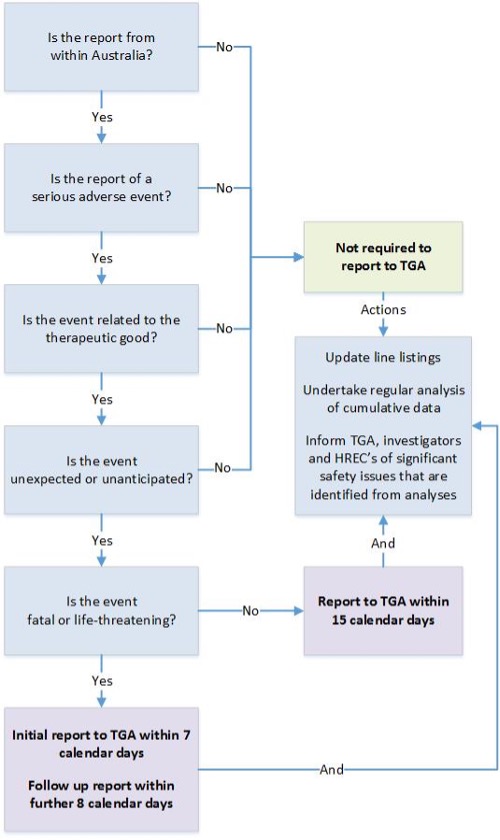
{kind=link}
This flowchart outlines the reporting requirements for SUSARs or USADEs of therapeutic goods in Australian clinical trials. It guides users through a series of questions to determine if a report is required to the Therapeutic Goods Administration (TGA).
Steps in the flowchart:
- Is the report from within Australia?
- If no, reporting to the TGA is not required.
- Is the report of a serious adverse event?
- If no, reporting to the TGA is not required.
- Is the event related to the therapeutic good?
- If no, reporting to the TGA is not required.
- Is the event unexpected or unanticipated?
- If no, reporting to the TGA is not required.
- Is the event fatal or life-threatening?
- If no, reporting to the TGA is required within 15 calendar days.
- If yes, an initial report to the TGA is required within 7 calendar days, followed by a follow-up report within 8 calendar days.
- Additional actions:
- Update line listings and undertake regular analysis of cumulative data for all reported events.
- Inform the TGA, investigators, and Human Research Ethics Committees (HRECs) of significant safety issues identified from analyses.
Significant safety issues including urgent safety measures
A significant safety issue (SSI) is a safety event that could adversely affect the safety of participants or materially impact on the continued ethical acceptability or conduct of the trial, for example by altering the risk-benefit balance of the trial. It is recommended that information regarding the trial sponsor’s proposed action, reports of any similar events internationally, and information as to the proposed action of other regulators is provided with notification of an SSI.
Example significant safety issues:
A serious adverse event that could be associated with the trial procedures and that requires modification of the conduct of the trial
A hazard to the patient population, such as lack of efficacy of an investigational medicinal product used for the treatment of a life-threatening disease
A major safety finding from a newly completed animal study (such as carcinogenicity)
Recommendations of the DSMB, where relevant for the safety of participants, such as an increase in frequency or severity of an expected adverse reaction.
A single case event such as a SUSAR or USADE may also meet the definition of an SSI. If this is the case it should be reported to us as both a SUSAR or USADE and an SSI. When the event is not an Australian SUSAR or USADE (i.e. from an overseas trial), report it solely as an SSI.
Example single case event that is also a significant safety issue:
Toxic epidermal necrolysis, agranulocytosis or hepatic failure in a participant that has led to an urgent safety measure.
SSIs requiring implementation of urgent safety measures (USMs) are of greatest concern to us. An USM is a measure required to be taken in order to eliminate an immediate hazard to a participant’s health or safety.
Example significant safety issue implemented as an urgent safety measure:
A chemotherapy trial requires an immediate dose reduction for all patients in response to an unacceptable level of toxicity seen in previously treated patients.
A temporary halt or early termination of a trial due to safety reasons would meet the definition of an SSI. The timelines for reporting to us would depend on whether the halt or termination required implementation of a USM.
In some trials, particularly those involving serious conditions (for example, intensive care trials), urgent actions or interventions may routinely be necessary, often as a result of the participant’s clinical condition. Where these events are managed in accordance with the protocol as routine care, they are not considered USMs. USMs are significant safety issues that result from the trial and are likely to result in a temporary halt or early termination, an immediate change to trial procedures or the addition of trial procedures. In these cases, the HREC is generally notified post hoc.
When an urgent safety measure occurs, where possible, the trial sponsor should notify us within 24 hours and in any case, no later than 72 hours of the measure being taken.
Refer to the NHMRC Guidance: Safety Monitoring and Reporting in Clinical Trials Involving Therapeutic Goods for safety reporting requirements for other stakeholders.
Sponsor reporting of significant safety issues to TGA (for trials conducted under the CTN or CTA schemes)
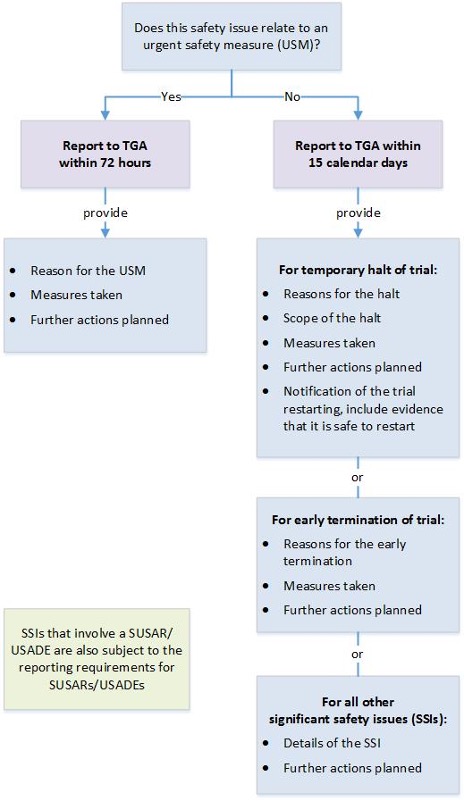
{kind=link}
This flowchart outlines the reporting requirements for significant safety issues of therapeutic goods in Australian clinical trials. It guides users through a series of questions to determine the appropriate reporting procedure to the Therapeutic Goods Administration (TGA).
Steps in the flowchart:
- Does this significant safety issue relate to an urgent safety measure (USM)?
- If yes, report to the TGA within 72 hours and provide:
- Reasons for the USM
- Measures taken
- Further actions planned
- If no, report to the TGA within 15 calendar days and provide:
- For temporary halt of trial:
- Reasons for the halt
- Scope of the halt
- Measures taken
- Further actions planned
- Notification of restarting the trial, including evidence of safety
- For early termination of trial:
- Reasons for early termination
- Measures taken
- Further actions planned
- For all other SSIs:
- Details of the SSI
- Further actions planned.
- For temporary halt of trial:
- If yes, report to the TGA within 72 hours and provide:
Additional note:
Significant safety issues (SSIs) that involve a suspected unexpected serious adverse reaction (SUSAR) or unanticipated serious adverse device effect (USADE) are also subject to the reporting requirements for SUSARs/USADES.
Other report types
Trial sponsors must keep records of all adverse events reported to them and are expected to maintain up-to-date tabulations or line listings of these events. This information should be made available on our request.
Trial sponsors should conduct an ongoing safety evaluation of the data they have received. If new safety information that meets the definition of an SSI is discovered it should be reported to us.
When data on ‘other adverse events’ are requested by us, initial presentation will be accepted in a tabular format, with further clinical details available at our request. As a minimum, this should include:
- participant identification codes,
- age,
- sex,
- name(s) of the therapeutic good(s) involved,
- dose and duration of treatment,
- nature of the reaction or event,
- condition being treated,
- potentially confounding factors and
- outcome.
Non-Australian SUSARs and USADEs
Trial sponsors are not required to routinely submit individual SUSARs or USADEs to us from outside Australia, even if a trial is ongoing at Australian sites.
However, trial sponsors are required to continually monitor the safety of their clinical development program and advise us of any SSIs that arise from analysis of overseas reports. This includes a temporary halt or early termination of an overseas trial.
In addition, any action with respect to safety which has been taken by another country's regulatory agency should be reported to us. This advice must include the basis for such action.
Trial sponsors must also inform the HREC(s) and investigator(s) of this information. Such information may be new and have an impact on the continued ethical acceptability of the trial, or may indicate the need for amendments to the trial protocol, including monitoring of safety. We also require that trial sponsors are able to provide to us promptly the clinical details of any individual overseas adverse event reports if requested.
Reports from post-market studies
For safety reporting requirements that apply only to post-ARTG registration studies where a medicine is being used in line with the product information or label indications, refer to Pharmacovigilance responsibilities of medicine sponsors.
For safety reporting requirements that apply only to post-ARTG inclusion studies carried out in accordance with the conditions of inclusion of a biological in the ARTG, label indications or product document indications refer to Biovigilance responsibilities of sponsors of biologicals.
Clinical trial phases and stages
Clinical trials of medicines and biologicals typically proceed through 'phases' of development whereas clinical trials of medical devices are more appropriately represented by ‘stages’.
The tables below provide a summary and comparison of the phases and stages of clinical trials involving the use of therapeutic goods. The table uses the objectives of the trial as the framework to assist trial sponsors to determine the phase or stage of the trial. The table should be considered as a guide only and is not intended to be exhaustive.
We acknowledge that clinical development pathways are becoming less rigid with respect to phase and that seamless adaptive trial designs and other cross-phase studies exist (for example, Phase Ib-II and Phase IIb-III).
Therefore, there may not be clear delineation between individual phases and stages. The basis for conducting clinical trials sequentially is that the results of prior studies should influence the plan of later studies.
However, the table below does not imply a fixed order of studies since the typical sequence may not be appropriate or necessary for the development of certain therapeutic goods.
Phase 0 has been introduced to assist in eliminating ineffective products early in the development process and is not considered to replace formal Phase I safety and tolerance studies.
Summary of clinical trial phases for medicines and biologicals
| Phase | Indicative number of participants | Objectives |
|---|---|---|
| Phase 0: Human pharmacology (micro-dosing) | 10-15 Involves dosing a limited number of humans with a limited range of doses for a limited period of time. | Assess pharmacokinetics: Gather preliminary data on pharmacokinetics and bioavailability to determine if the drug behaves as expected from preclinical studies ‘micro-dosing’ studies. |
| Phase I: Human pharmacology | 10-100 May involve the first administration to humans, usually to small numbers of healthy volunteers or to patients. | Safety and tolerance:
|
| Phase II: Therapeutic exploratory | 100-300 May be undertaken in a larger group of human patients (several hundred). | Efficacy and safety:
|
| Phase III: Therapeutic confirmatory | 300-3000 Usually involve a large group of patients (from several hundred to several thousand). | Safety, efficacy or effectiveness:
|
| Phase IV: Therapeutic use | 1000’s | Post marketing surveillance or resolution of treatment uncertainties:
|
Summary of clinical trial phases for medical devices
| Stage | Indicative number of participants | Objectives |
|---|---|---|
| Pre-market pilot | 10-30 Usually involves a small group of human patients | Exploratory investigations to determine preliminary safety and performance information to plan design modifications or provide support for a future pivotal study. (Includes first in human and feasibility studies or proof of concept) |
| Pre-market pivotal | 100’s | Confirmatory investigations to evaluate performance and safety for a specified intended use to satisfy pre-market regulatory requirements |
| Post-market | 1000’s |
|
The phases and stages of clinical trials summarised in the tables above are described extensively in other documents; however, in relation to the safety of Australian participants, early phase trials; particularly first in human trials pose the greatest risk.
The following section provides additional considerations for early phase trials and also links to further information.
Early phase trials
Early phase trials are no longer defined as traditional Phase I trials. Early phase trials can be broadly defined as non-therapeutic, exploratory trials in human participants who may be healthy volunteers or have a specific disease.
Over recent years, the conduct of early phase trials has evolved. Increasingly, trial sponsors are submitting early phase trials to HRECs that combine a number of different study parts within an integrated trial protocol. Common activities and objectives of early phase clinical trials using integrated trial protocols (human pharmacology) are outlined in the table below.
Early phase clinical trials using integrated trial protocols (human pharmacology)
| Initial studies | Follow-up studies |
|---|---|
Possibly pharmacodynamics (PD) |
|
Similar activities and objectives may be applied to early phase trials for biologicals, with additional consideration of the potential risk posed by the biological to patients, medical personnel and the population as a result of the origin of the cells or tissue, the manufacturing process, or the route of administration. Additional aspects of integrated trial protocols may include:
- requirement for concomitant medication, for example immunosuppressive regimens, for effect
- monitoring of viability, proliferation or differentiation, migration and persistence
- specific potential toxicities (for example, immune responses, infections, malignant transformation).
There are several clinical and non-clinical guidelines that should be considered by HRECs, trial sponsors and manufacturers in the context of early phase trials including:
- Non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals (ICH M3 (R2))
- ICH guideline M3 (R2) - Questions and Answers
- EMEA/CHMP Guideline on human cell-based medicinal products (EMEA/CHMP/410869/2006)
- EMA/CAT Reflection paper on stem cell-based medicinal products (EMA/CAT/571134/2009)
- EMEA/CHMP Note for guidance on the quality, preclinical and clinical aspects of gene transfer medicinal products (CPMP/BWP/3088/99)
As outlined above, first in human trials have the greatest element of uncertainty in relation to risk. Obtaining appropriate nonclinical data and properly considering this information is critical to safety. The European Union Guideline: Strategies to Identify and Mitigate Risks for First-In-Human Clinical Trials with Investigational Medicinal Products includes recommendations that incorporate the findings from investigations following overseas clinical trial incidents to appropriately manage the risk posed. It also provides guidance on appropriate sentinel dosing.
There are four key recommendations that trial sponsors should consider when conducting early phase trials:
- Better identification of factors influencing risk
- mode of action (for example, targeting multiple pathways, having long lasting effects)
- nature of target (for example, ‘down-stream’ effects of target and ‘off target’ effects)
- relevance of animal species and models
- findings in non-clinical safety studies
- Careful dosing selection
- use of minimum anticipated biological effect level (MABEL) and pharmacologically active dose (PAD) and not just no observed adverse effect level (NOAEL)
- better justification of dose escalation
- maximum dose should take into account target saturation (for example, where the intended therapeutic effect is linked to enzyme inhibition, when complete inhibition is achieved)
- Better justification for and planning of integrated protocols
- scientifically justify overlap of SAD and MAD parts of the study
- ensure sufficient space between sub-studies
- Careful selection of participant group and development of other treatment precautions
- justify the decision to include healthy volunteers or patients
- ensure adequate time period between doses within cohorts and between cohorts.
To minimise any issues, including the risk of adverse events in early phase clinical trials, we recommend that HRECs, trial sponsors and manufacturers utilise individuals with adequate expertise to assess and manage higher risk early phase trials.
In response to the changing environment, some jurisdictions are developing frameworks for early phase clinical trials which includes the appointment of specialist early phase trial HRECs. The NHMRC has assessed and certified the ethics review processes of certain institutions under the National Certification Scheme. A list of HRECs with certified ethics review processes is available on the NHMRC website. We recommend, at a minimum, that first in human trials are reviewed by a HREC on this list with a certification category relevant to the phase of the trial.
Medical device stages
A new device is often subject to extensive preclinical testing through engineering analysis and testing, computational simulation, biocompatibility testing including immunogenicity and carcinogenicity testing and, in appropriate instances, animal testing.
The invasive nature of many medical devices precludes initial testing in healthy volunteers, necessitating the use of animal model testing. Initial clinical testing of devices usually involves a pilot study in small groups of patients.
If the feasibility of the concept is proven, larger studies with well-designed trial protocols and a sound statistical basis are undertaken. Studies may also be carried out to confirm the performance and safety of changes in design or material of a device or to assess the device's performance against new clinical indications.
The clinical safety and performance of many devices depends largely on the experience and training of the clinician using the device. These are important considerations when assessing a clinical trial application. Implantable devices also require special consideration. A surgical procedure in itself poses risk to a patient; and in the event of a device failure, a second surgical procedure may be required to remove a faulty experimental device.
We recommend that HRECs, trial sponsors and manufacturers utilise individuals with adequate expertise (such as clinical experience using similar devices) to assess and manage higher risk early stage trials to minimise any issues, including the risk of adverse events, as with early phase clinical trials with medicines. This includes the technical considerations for medical device design and preclinical evaluation.
The Essential Principles checklist set out the requirements relating to the safety and performance characteristics of medical devices. When applying for inclusion in the ARTG, evidence from a clinical trial will be required to demonstrate that a manufacturer has validated the design of the device, and to provide objective evidence to substantiate compliance with some of the Essential Principles.
Advertising ‘unapproved’ therapeutic goods
It is an offence to advertise any therapeutic good that has not been included in the ARTG under the Therapeutic Goods Act 1989 (section 42DL(1)). While the trial sponsor would be able to promote their trial in the public domain, they could not specifically mention the name of the therapeutic good being used in the trial.
Section 42DL(1) also notes a person may not publish or broadcast an advertisement about therapeutic goods that contains a prohibited or restricted representation, or that contains a statement referring to goods, or substances or preparations containing goods, included in Schedule 3, 4 or 8 to the current Poisons Standard.
An ‘advertisement’, in relation to therapeutic goods, includes any statement, pictorial representation or design, however made, that is intended, whether directly or indirectly, to promote the use or supply of the goods.
A person must not make a claim that they can arrange the supply of goods not in the ARTG (Section 22(6), 32BJ(4)(a) and 41MM of the Therapeutic Goods Act 1989).
Any advertisement for a clinical trial should be approved by the HREC reviewing the trial as specified under section 5.2.23 of the National Statement and section 4.4.1 of the Guideline for Good Clinical Practice.
See therapeutic goods advertising in Australia for more information.
Manufacturing
Trial sponsors undertake the ultimate responsibility for all aspects of the clinical trial including the quality of investigational products.
Different parts of the Therapeutic Goods Act 1989 apply to the manufacture of medicines, biologicals and other therapeutic goods (Part 3-3 of Chapter 3) and to the manufacture of medical devices (Part 4-3 of Chapter 4). In addition, manufacturers must comply with any relevant state and territory legislation.
Manufacturing medicines and biologicals for use in a clinical trial
Manufacture in Australia
Australian facilities participating in the manufacture of a medicine or biological for use in a clinical trial must hold a manufacturing licence issued by us. However, certain persons or goods are exempt from this requirement. Even if a pilot facility is dedicated for the development of dosage forms and new products, and is not used for the manufacture of saleable product, it is still subject to inspection and licencing by us unless an exemption applies.
Even if a pilot facility is dedicated for the development of dosage forms and new products and is not used for the manufacture of saleable product, it is still subject to inspection and licencing by us unless an exemption applies.
Manufacturers need to comply with:
- the manufacturing principles specified in Australian legislation
- relevant Therapeutic Goods Orders
- relevant default standards
- any conditions applying to the inclusion of the goods in the ARTG (if included in the ARTG).
Further information regarding obtaining a manufacturing licence can be found at Australian manufacturing licences and overseas GMP certification.
Manufacturing principles
Good manufacturing practice (GMP) is applied to the manufacture of investigational medicinal products or investigational biologicals to ensure those products are fit for their intended use and do not place participants at risk due to inadequate quality, safety or efficacy.
Section 5.13.1 of the Guideline for Good Clinical Practice states that the trial sponsor must ensure that investigational products, including active comparators and placebo, are manufactured in accordance with any 'applicable GMP'. The applicable GMP in Australia is specified in the current Therapeutic Goods (Manufacturing Principles) Determinations. These principles require manufacturers to demonstrate that manufacturing practices comply with a relevant code of GMP. There are different codes of GMP depending on the type of therapeutic good:
- Medicines and biologicals that comprise or contain live animal cells, tissues or organs (PIC/S Guide to Good Manufacturing Practice for Medicinal Products)
- Part I of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products applies to the manufacture of finished investigational medicinal products (including placebos):
- In particular, Annex 13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products specifically applies to the manufacture of investigational medicinal products that are required to comply with the manufacturing principles. Note: refer to Annex 13 for information regarding when reconstitution is not subject to manufacturing authorisation.
- Part II of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products applies to the manufacture of active pharmaceutical ingredients:
- Section 19 outlines the GMP requirements that apply to the manufacture of active pharmaceutical ingredients for use in clinical trials.
- The Annexes of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products may also apply where relevant to the specific manufacturing operations. For example, Annex 1 applies to the manufacture of sterile products and active pharmaceutical ingredients.
- Human blood, blood components, haematopoietic progenitor cells (HPCs) and biologicals that comprise, contain or are derived from human cells and tissues (The Australian Code of Good Manufacturing Practice for Blood and Blood Components, Human Tissues and Human Cellular Therapy Products)
PIC/S has also published an Aide-Memoire on GMP particularities for clinical trial products (PI 021-2) in 2007, which acknowledges that investigational medicinal products should be produced in accordance with the principles and guidelines of GMP for medicinal products. This also notes that there may be added risks to participants compared to patients treated with ‘marketed’ products.
Departure from Annex 13
In exceptional circumstances it may not be possible to meet the requirements of Annex 13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products for labelling investigational products.
In this case, the trial sponsor must contact us if they wish to request a departure from the requirements of Annex 13. Sub-clause 3 of the Therapeutic Goods (Manufacturing Principles) Determination 2018 provides the criteria that we must be satisfied with if an alternative procedure to the procedure or requirement set out in the PIC/S Guide to Good Manufacturing Practice for Medicinal Products is adopted.
The PIC/S Guide to Good Manufacturing Practice for medicinal products recognises that there are acceptable methods, other than those described in the guides that are capable of achieving the principles of GMP established in the guides. The trial sponsor may choose to use an alternate method if a risk assessment demonstrates that the alternative approach provides the same or greater quality and safety.
However, a formal risk management plan must be documented and included with the trial documentation if the requirements are not met. Annex 20 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products outlines approaches to be used and tools which a trial sponsor may choose to use when applying a formal quality risk management approach.
Manufacture overseas
We are unable to enforce GMP standards applicable in Australia to the manufacture of investigational products in another country. Therefore, we recommend that the trial sponsor ensures that they have a thorough understanding of the principles of GMP and ensure that the manufacture of medicines and biologicals for use in clinical trials is in accordance with the principles of GMP.
We recommend that the trial sponsor ensures that the GMP licensing for other countries adheres to the principles of GMP as set out in the PIC/S Guide for Good Manufacturing Practice for Medicinal Products or the Australian code of Good Manufacturing Practice for blood and blood components, human tissues and human cellular therapy products in order to assess the quality of the product.
Labelling medicines
The labelling therapeutic goods orders TGO 69, TGO 91 and TGO 92 do not apply to investigational medicinal products. Instead, labelling requirements are specified in Annex 13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products. This document outlines the information to be included on labels, unless its absence can be justified, for example, use of a centralised electronic randomisation system.
Complete details on primary and secondary packaging and labelling requirements can be found in Annex 13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products. However, some items have been discussed further below.
Notes on clauses in Annex 13
| Clause | Notes |
|---|---|
| 26(a) | The main contact details for information on the product, clinical trial and emergency unblinding must be an Australian contact. |
| 26(b) | For closed blinded trials, the labelling should include a statement indicating “placebo or [name/identifier] + [strength/potency]”. |
| 26(d) | The trial reference code used should identify the particular trial site, unless provided elsewhere or its absence can be justified. This is the case even for trials involving multiple sites. |
| 26(d) | The trial reference code used should identify the Australian trial sponsor, unless provided as the main contact (clause 26(a)) or its absence can be justified. It is not sufficient to identify only an overseas trial sponsor in the case of a multinational trial. |
| 26(f) | The name of the principal investigator should appear on the label unless already included in a trial reference code or unless its absence can be justified. |
| 27 | Allows for a participant to be provided a leaflet or card that contains the address and telephone number of the main contact for information on the product, clinical trial and for emergency unblinding. The participant must be instructed to keep this in their possession at all times. |
| 32 | 'Certain characteristics' refers to non-commercial clinical trials performed by researchers without the participation of the pharmaceutical industry. These trials are often performed with registered (or listed) products that are obtained from the market for use in a clinical trial. The requirements in this clause relate to the way these products are to be labelled. |
| 33 | We do not allow labels to omit an expiry, use-by or re-test date. Trial sponsors must comply with the requirements of this clause. |
Labelling is a manufacturing step under the Therapeutic Goods Act 1989. However, an exemption from the requirement to hold a manufacturing licence may apply to certain persons identified within Schedule 8 of the Therapeutic Goods Regulations 1990, to allow relabelling of the investigational medicinal product with name and address of the new sponsor. See Manufacturing exemptions.
If there is a change of Australian trial sponsor, the clinical trial medication should be re-labelled appropriately with the details of the new trial sponsor at the time of transfer.
Labelling and centralised electronic randomisation systems
The labelling requirements of Annex 13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products must generally be met, however, certain information may be omitted from the label where centralised electronic randomisation systems (CERS) are used.
When CERS are used, and investigational medicinal products are:
- distributed to participants to administer at home, the label should contain:
- sufficient information so that any person can readily identify the product if required (such as in an emergency), and
- the main contact for product information, clinical trial and emergency unblinding, and
- directions for use, and
- storage conditions and expiry, use-by or re-test date
- administered by dedicated trial staff and no additional investigational medicinal product is retained by participants, it may be possible to:
- omit information (except the expiry date) from the labels in a readable format, provided that alternative means of accessing, verifying and retaining relevant information is provided by the CERS.
The decision to omit certain information should be made according to risk management principles and information should be checked immediately before administering the investigational medicinal product.
Labelling biologicals
Investigational biologicals must comply with the labelling requirements set out in the Australian code of Good Manufacturing Practice for blood and blood components, human tissues and human cellular therapy products. The exception is biologicals that comprise or contain live animal cells, tissues or organs, which must comply with Annex 13 of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products.
Manufacturing medical devices for use in a clinical trial
There are no specific requirements prescribed in the Therapeutic Goods (Medical Devices) Regulations 2002 for the manufacture of investigational medical devices for use in clinical investigations, other than those that are incidental to the demonstration of compliance with the Essential Principles in relation to the construction of the device.
For medical devices, the mechanism for a manufacturer to apply a conformity assessment procedure has been defined for an investigational device under Regulation 3.10 of the Therapeutic Goods (Medical Devices) Regulations 2002.
However, there is currently no requirement in the special conformity assessment procedures (Part 7 of Schedule 3 to the Therapeutic Goods (Medical Devices) Regulations 2002) for the manufacturer of such a device to apply a quality management system.
In practice, the regulation of the manufacture of medical devices is implemented by:
- the application of ISO 13485 – Medical Devices, Quality Management Systems Requirements for Regulatory Purposes
- certification by an independent third party.
Manufacturing records and evidence of compliance
We acknowledge that a start-up medical device company may not yet have received third party certification for their quality management system at the time they are ready to start early pilot studies of their product.
However, it is expected that design, development, control of raw materials, environmental controls, quality controls and record keeping in the manufacture of medical devices for investigational purposes is conducted in accordance with the requirements of this standard; and that the manufacturer is actively preparing for certification.
An established medical device company that is manufacturing investigational medical devices should already have the appropriate conformity assessment certification, which includes the requirements of ISO 13485. We recommend that the manufacture of invasive investigational medical devices is undertaken by a certified manufacturer.
Sponsors of ‘unapproved’ medical devices that are intended for use in a clinical trial should ensure that the manufacturer of the medical device has evidence to substantiate compliance with the Essential Principles, that are relevant to their medical device, and may be found in Schedule 1 to the Therapeutic Goods (Medical Devices) Regulations 2002 (apart from those aspects of the Essential Principles that require the generation of clinical evidence).
Evidence of compliance should be documented and available for review by those that have a role in the approval of the clinical trial (for example, the HREC).
Technical documentation
In addition to the manufacturing records for the investigational medical device, a technical file, or design dossier, should be established for this purpose. As a general guide, the technical documentation that is held in this file by the manufacturer should include:
- a general description of the medical device
- the intended purpose of the medical device
- drawings of the design of the medical device, including components, subassemblies or circuits
- descriptions or explanations that are necessary to understand the drawings of the medical device and its intended purpose
- a statement indicating whether or not the medical device incorporates, or is intended to incorporate, as an integral part, a substance that may be considered to be a medicine
- a statement indicating whether or not the medical device, other than an IVD medical device, contains tissues, cells or substances of animal origin that have been rendered non-viable, or tissues, cells or substances of microbial or recombinant origin
- for an IVD medical device—a statement indicating whether or not the device contains viable tissues, cells, or substances of human or animal origin
- a description of any sterilisation processes used in the manufacture of the medical device
- details of any standards used in relation to the manufacture of the medical device
- design calculations, risk analyses and technical tests or any other investigations carried out on the medical device to establish compliance with the relevant Essential Principles
- where the medical device is to be used in conjunction with (for example, connected to) other medical devices, results of tests demonstrating the safety and performance of each medical device is acceptable
- a copy of the information to be provided with the medical device.
The Essential Principles checklist is a useful tool when preparing a cross-reference or index to the evidence of compliance. There are many international standards relevant to the design, testing and manufacture of medical devices that are commonly applied to demonstrate compliance with the Essential Principles. Due to the alignment of Australian medical device requirements with those of Europe, the European harmonised standards for medical devices may be used as a reference.
Although a quality management system is not strictly required for the manufacture of an investigational medical device, some aspects related to the manufacturing (construction) of medical devices are specified in the Essential Principles. Evidence supporting compliance must be available prior to use in a clinical trial.
Labelling medical devices
The requirements for medical device labelling are specified in clause 13.3 of Schedule 1 to the Therapeutic Goods (Medical Devices) Regulations 2002 (Essential Principles information to be provided with a medical device). This includes investigational devices.
The ISO 14155 states that the investigational device, the instructions for use or the packaging should indicate that the investigational device is exclusively for use in a clinical investigation.
Some of the principles outlined in Annex 13 of the PIC/S Guide for Good Manufacturing Practice for Medicinal Products, may also have relevance to investigational medical device labelling.
Manufacturing exemptions
Manufacturing exemptions for medicines and biologicals
Certain exemptions from Part 3 of Chapter 3 of the Therapeutic Goods Act 1989 are outlined below.
Therapeutic goods that are not medical devices and are manufactured in Australia may only be manufactured by persons who are licensed to manufacture (or to carry out a step in the manufacture of such goods at licensed premises) unless either the goods or the person responsible for the manufacture are identified in the legislation to be exempt.
The Guideline for Good Clinical Practice states that the trial sponsor should ensure that investigational products are manufactured in accordance with any 'applicable GMP' (see Manufacturing principles above). Therefore, we still recommend compliance with the principles of the PIC/S Guide to Good Manufacturing Practice for Medicinal Products (in particular Annex 13) and the Australian Code of Good Manufacturing Practice for Blood and Blood Components, Human Tissues and Human Cellular Therapy Products where an exemption applies.
Trial sponsors should ensure that they have thorough understanding of the principles of GMP in order to assess the quality of the therapeutic goods used in the trial. GMP compliance of the manufacturer is recommended to support the acceptance of clinical trial data overseas. We recommend the trial sponsor seeks independent legal advice to ensure compliance with the legislation. This may include the applicable legislation of the jurisdictions in which the trial material is used.
Therapeutic goods exempt from the operation of Part 3-3 of the Therapeutic Goods Act 1989 include goods prepared for the initial experimental studies in human volunteers as specified in item 1, Schedule 7 to the Therapeutic Goods Regulations 1990.
Refer to Schedule 7 of the Therapeutic Goods Regulations 1990 for the full list of exemptions.
In practice, initial experimental studies in human volunteers means first-in-human trials. This will generally, but not always, be phase 0 and I trials (human pharmacology trials).
No such exemption exists for therapeutic goods used in other phase studies. Therefore, the manufacture of all other investigational medicinal products and biologicals in Australia is subject to inspection and licensing by us (unless a separate exemption applies).
Manufacturers of investigational products that are not initial experimental studies in human volunteers must hold a valid TGA licence that specifically authorises that site for the manufacture of clinical trial products in Australia (unless a separate exemption applies).
Persons exempt from the operation of Part 3-3 of the Therapeutic Goods Act 1989 include:
- pharmacists who manufacture therapeutic goods in a pharmacy where the pharmacist practices and the pharmacy is open to the public and the goods are supplied from those premises and meet the requirements of item 2, Schedule 8 to the Therapeutic Goods Regulations 1990
- pharmacists employed by a public hospital or public institution who manufacture therapeutic goods for supply in hospitals and public institutions in the same state or territory and meet the requirements of item 3, Schedule 8 to the Therapeutic Goods Regulations 1990
- a person who applies supplementary labelling to a manufactured product, where the supplementary label contains only a name and address, the registration or listing number of goods, or the biological number of a biological as specified in item 5, Schedule 8 to the Therapeutic Goods Regulations 1990.
Refer to Schedule 8 of the Therapeutic Goods Regulations 1990 for the full list of exemptions.
Manufacturing exemptions for medical devices
Full exemption from the penalties for non-compliance with the Essential Principles, is not normally available for any medical device, unless the medical device is for emergency use and has been granted an exemption under section 41GS of the Therapeutic Goods Act 1989.
However, it is recognised that compliance with the Essential Principles that require clinical evidence, cannot be demonstrated until the clinical trial has been completed.
Importing
Import restrictions for therapeutic goods
Restrictions may be placed on the import of therapeutic goods for clinical trials. Some of these restrictions have been outlined below (this list is not exhaustive).
- therapeutic goods legislation (see below)
- Customs (Prohibited Imports) Regulations 1956 and – a licence and or permit to import or export may be required for substances controlled under this legislation
- Biosecurity Act 2015 – permission may be required prior to importing any material of biological origin (human, animal, plant or microbial)
- Environment Protection and Biodiversity Conservation Act 1999 – permission may be required prior to importing endangered species
- Gene Technology Act 2000 – permission may be required prior to importing genetically modified organisms
- state and territory requirements (contact the relevant state or territory department of health)
- requirements of the legislation of the originator country.
Importing therapeutic goods for use in clinical trials in Australia
The Therapeutic Goods Act 1989 prohibits import into or supply in Australia of ‘unapproved’ therapeutic goods for use in humans unless they are included in the ARTG or otherwise the subject of an exemption, approval or authority.
Item 1 of Schedule 5A to the Therapeutic Goods Regulations 1990 and item 2.1 of Schedule 4 to the Therapeutic Goods (Medical Devices) Regulations 2002 provide an exemption for therapeutic goods to be imported and held prior to supply in a clinical trial subject to conditions.
The imported therapeutic goods must be held under the direct control of the sponsor (importer) until the goods are the subject of a notification under the CTN scheme or an approval under the CTA scheme. Until such time, the goods must be kept in a warehouse or a properly secured area under the control of the importer in accordance with the relevant exemption.
There is no requirement for the CTN or CTA process to have been completed prior to importation of the clinical trial goods. However, approval through the CTA scheme or notification through the CTN scheme needs to have occurred prior to supply of the goods to the trial sites.
Other conditions relevant to the exemption include keeping records relating to the source and supply of the goods. These records should be provided to us if requested.
Importing therapeutic goods for use in clinical trials
-
Sponsor (importer)
Checks import restrictions and applies for any relevant permissions
-
Sponsor (importer)
(Item 1 of Schedule 5A of the Regulations)
Imports clinical trial goods into Australia
-
Sponsor (importer)
(Item 1 of Schedule 5A of the Regulations)
Holds clinical trial goods under direct control
-
Sponsor (importer)
Supplies goods to trial sponsor under CTN exemption or CTA approval
-
Clinical trial sponsor
Supplies therapeutic goods to participants in clinical trial (subject to HREC and institutional authorisations)
Six monthly reports
A sponsor of therapeutic goods in relation to an exemption under section 18, 32CA or 41HA or approval under section 19, 32CK or 41HB of the Therapeutic Goods Act 1989 must provide a report to us every six months. Regulation 47B of the Therapeutic Goods Regulations 1990 outlines the details required in such reports.
Retrieval, destruction and export of unused clinical trial material
The Guideline for Good Clinical Practice outlines that trial sponsors need to maintain a system for the retrieval and disposition of investigational product(s) and for the documentation of this disposition.
There are provisions under the therapeutic goods legislation that allow the export of therapeutic goods for destruction. Under item 2 of Schedule 5 to the Therapeutic Goods Regulations 1990 and item 1.2 of Schedule 4 to the Therapeutic Goods (Medical Devices) Regulations 2002 ‘unapproved’ therapeutic goods may be exported from Australia if they:
- are not for commercial supply
- do not contain a substance the exportation of which is prohibited under the Customs Act 1901
- are not intended for use in clinical trials on humans.
Therefore, if the above conditions apply then the goods may be exported for destruction without any requirement for export approval from us. The Office of Drug Control provides advice on substances that may be prohibited exports.
Trial sponsors are also advised to contact the relevant department in the destination country to determine requirements.
Footnotes
- The use of the expression ‘unapproved’ in this handbook refers to the international expression ‘approval for general marketing’. It does not refer to the absence of any express statutory approval under the therapeutic goods legislation, but rather the absence of any registration, listing or inclusion of the therapeutic goods in the ARTG.
Page history
Content migrated to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Minor updates to include CTA information.
CTX name change to CTA.
Updated hyperlinks.
- minor updates to text and hyperlinks
- updated the reporting methods for SUSARs and USADEs. Removed recommendation to submit IB&P and trial protocol with individual SUSARs and USADEs
- removed the information requiring importers to destroy or return unapproved goods if they are not used within 12 months of authorisation (legislative update)
- further clarification provided on checking overseas legislative requirements for overseas trials, compliance with National statement, managing conflicts of interest, and transferring sponsorship.
Restructured and brought up to date to reflect current practices and meet government accessibility requirements.
Replaces:
- Access to unapproved therapeutic goods - Clinical Trials in Australia (October 2004)
- Human Research Ethics Committees and the Therapeutic Goods Legislation (June 2001)
- Australian Clinical Trials Handbook (March 2006).
Original publication.
The Australian Clinical Trial Handbook is a simple and practical guide to the conduct of clinical trials to International standards of Good Clinical Practice (GCP) in the Australian context.
Content migrated to new 'Guidance' content type:
- Consistent ‘Purpose’ heading.
- ‘Legislation’ section to clearly show which laws the Guidance relates to.
- ‘Page history’ section replaces document version history.
- New page navigation features.
- Updated page summaries.
- Complex images include long descriptions.
- New ‘Save as PDF’ feature.
Minor updates to include CTA information.
CTX name change to CTA.
Updated hyperlinks.
- minor updates to text and hyperlinks
- updated the reporting methods for SUSARs and USADEs. Removed recommendation to submit IB&P and trial protocol with individual SUSARs and USADEs
- removed the information requiring importers to destroy or return unapproved goods if they are not used within 12 months of authorisation (legislative update)
- further clarification provided on checking overseas legislative requirements for overseas trials, compliance with National statement, managing conflicts of interest, and transferring sponsorship.
Restructured and brought up to date to reflect current practices and meet government accessibility requirements.
Replaces:
- Access to unapproved therapeutic goods - Clinical Trials in Australia (October 2004)
- Human Research Ethics Committees and the Therapeutic Goods Legislation (June 2001)
- Australian Clinical Trials Handbook (March 2006).
Original publication.
The Australian Clinical Trial Handbook is a simple and practical guide to the conduct of clinical trials to International standards of Good Clinical Practice (GCP) in the Australian context.